A szarvas emblematikus, kultikus lény, amelyet tisztelet és csodálat övez. Újkőkori barlangrajzok, 5-6000 éves ékírások, művészi szobrocskák őrzik alakját. A középkor „királyi vadja”, rengeteg szépirodalom hőse az ókortól máig, megjelenése gazdag a magyar irodalomban is. Több nép mitológiájának szereplője, a magyar mitológiának is központi alakja a Csodaszarvas, egyházi és világi zeneművek dicsőítik, képzőművészeti alkotások, fotóművészet tárgya, világhírű természetfilmek és művészfilmek szereplője (például Homoki Nagy István: Gyöngyvirágtól lombhullásig; Enyedi Ildikó: Test és lélek). A világelső trófeákról sem feledkezhetünk meg, amelyek itt a Kárpát-medencében „teremnek”. A Dél-Dunántúlon és Gemencen él a világ legkiválóbb gímszarvas populációja. A szarvasok genetikai kutatását, melynek eddigi csúcsteljesítménye a CerEla1.0-kutatás, nem kis mértékben motiválta az a mintegy 20 ezer év, amely a szarvasokat az emberi kultúrához köti.
A genetika újévi híre 2018. január másodikán egy igen érdekes cikkben jelent meg. Ebben beszámoltak a Mezőgazdasági Biotechnológiai Központ (MBK, NAIK, Gödöllő), a Kaposvári Egyetem állattenyésztési, az ELTE genetikai, és a SOTE 1. Belgy. Klinika MSc és PhD programjainak összefogásával lefolytatott program sikeréről: meghatároztatott a gímszarvas DNS-ének szekvenciája és elkészült a gímszarvas teljes genomjának leírása, amely CerELa1.0 névre kereszteltetett. Ez nemzetközileg is jelentős felfedező kutatás: a gímszarvas genomjának első leírása a világon. A CerEla1.0 program egy hosszú út végét jelzi, amely 1998-ban indult, koncepcióját Orosz László dolgozta ki, az állattenyésztési és vadgazdálkodási hasznosítást Horn Péter, a klinikai irányt Lakatos Péter jegyezte. A CerEla1.0-ról beszámoló tudományos közlemény a rangos Molecular Genetics and Genomics (MGG) tudományos folyóiratban 2018. január 2-án jelent meg online (https://doi.org/10.1007/s00438-017-1412-3). Az MGG-ről megemlítendő, hogy a világ elsőként alapított genetikai folyóirata (1908, Springer), s a magyar molekuláris genetikai kutatások (SZBK, Szeged, 1970-nes évektől) itt szereztek először széleskörű nemzetközi elismerést.
Hatalmas információmennyiség
A CerEla1.0. leírja a 33 gímszarvas autoszóma, valamint az X és Y nemi kromoszómák DNS szekvenciáját, a centromeronok helyzetét, közel 20 ezer fehérjét kódoló gént, továbbá a riboszóma RNS (rRNS), transzfer RNS (tRNS) és kis-RNS (sRNS) géneket. Tartalmazza a gének részletes annotációját, azaz hogy milyen funkciót látnak el, mi a megfelőjük (ortológok) más emlősfajokban, megjeleníti a gének szerkezetét (szabályozó régiók, exon- és intronszerkezet, 5’ és 3’ nem transzlálódó régiók, UTR-ek stb.), az ismétlődő, repetitív szekvenciákat és mikroszatellitákat, mozgó genetikai elemeket, transzpozonokat és LTR-elemeket. Mindezen genetikai elemek helyzete, sorrendje, távolsága pontosan kikereshető a kromoszómák mentén a DNS-szekvenciában. A CerEla1.0 lehetőséget adott 2,8 millió heterozigóta SNP-hely meghatározására (single nucleotide polimorphysm, bázispár különbségek az adott egyed apai és anyai eredetű, homológ kromoszómái között) és 365 ezer DNS-deléció és inszerció azonosítására a szarvasgenom mentén (deléció: egy DNS szakasz kiesése, inszerció: egy DNS szakasz beékelődése a „vad típusú” DNS szekvenciába, közös néven indel-ek). Megjegyzendő, hogy az emlősök – így a gímszarvas is – ugyanazokat a géneket hordozzák, amelyek DNS-szekvenciája nagyrészt megegyezik, pl. a szarvas és az ember egymásnak megfelelő génjei (az ortológok) 90-95 %-ban megegyeznek. Saját kutatásaink pedig azt mutatták, hogy a szarvas és a szarvasmarha esetében az egyezés 98-100% a gének kódoló részein és 90% körüli a promóter/ szabályozó régiókban. Azt, hogy a fajok különböznek, alapvetően a gének szabályozásában és a génműködési hálózatok különbségeiben kell keresni. Az emlősök, így az ember, vagy gímszarvas haploid genomjában a kromoszómák összesen kb. 3 milliárd DNS bázispárt (3Gbp) tartalmaznak, ami sorba rakva mintegy 1 méter hosszú DNS fonal lenne, azaz ennyi DNS van egy petesejtben, illetve egy spermiumban. A megtermékenyített petesejtben (zigótában), illetve a diploid testi sejtekben kétszer ennyi. (Itt most eltekintettünk a mtDNS ről, a mitokondriumok DNS-éről, amelyek az energia háztartásért felelős géneket hordozzák, s amelyek a petesejt citoplazmáján keresztül jutnak a megtermékenyítés következményeként a zigótába, majd a testi sejtekbe. A mtDNS mindig anyától öröklődik utódjára – ez az „anyai öröklés” jelensége.) A CerEla1.0 értékes adatokkal bővítette a kérődzők genom/kromoszóma evolúciójáról (a kariológiai evolúcióról) alkotott képet is. Az őskérődzők (Pecora, a mai szibériai pézsmaszarvas áll közel ehhez az ősi állapothoz) az oligocén kor elején 30 millió éve jelentek meg, leszármazottaik a szarvasfélék (Cervidae család, 50 ma élő faj, szarvasok, őzek stb.) ősei 23 millió, a tülkös szarvúak (Bovidae család, 150 mai faj, tulkok, juhok, antilopok stb.) ősei valamivel később, 20 millió éve alakultak ki. A Cervidae és a Bovidae evolúció millió évei során a kifejlődött fajokban az eredeti Pecora kromoszómakészlet átrendeződött, ősi kromoszómák széthasadtak kettőre, mások összeolvadtak eggyé (fúzió), ősi kromoszómaszakaszok megfordultak a kromoszómákon belül (inverzió), esetenként áthelyeződtek (transzlokáció) egyik kromoszómáról a másikra. Mindezen változásokat őrzi a ma élő fajok genomja és precízen kiolvashatók a CerEla1.0 DNS-szekvenciából is.
Összehasonlító géntérképezés
A következő fejezetben lesz szó arról, hogy milyen lépésekben épült fel a CerEla1.0. Ez azonban a genetika két alapvető törvényének alkalmazása nélkül nem lett volna megvalósítható. Az egyik a klasszikus géntérkép és a kromoszóma kolinearitása, a másik az összehasonlító géntérképezés elve. A klasszikus géntérképezés az úgynevezett crossing-over törvények (pl. 2 pontos, 3 pontos elemzés) alkalmazásán és a genetikai rekombináció kimutatásán alapul. A rekombináció és gyakorisága (R) irányított keresztezések után, mint pl. a tesztelő keresztezés (teszt-cross), az utódokból olvasható le. Minél távolabb van két gén a kromoszómán (kapcsolási csoportban), annál nagyobb a rekombináció gyakorisága. Ez a gyakoriság azonban nem egyenesen arányos a rekombinálódó gének távolságával, mivel csakis a páratlan számú crossing-over eseményeket tudjuk kimutatni a teszt-cross módszerrel. Ezek az események jelenítik meg számunkra a genetikai rekombinációt. A gének közötti távolság és a gének közötti rekombináció gyakoriságának összefüggését a genetikai térképezési függvény, a Haldane-függvény írja le: Ra-b= ½ (1-e-2d), ahol R az a és b pontok/gének között a rekombináció gyakorisága, d az a és b pontok/gének távolsága Morgan (M) egységekben kifejezve. 1 M azon két pont/gén távolsága, amelyek között 1, azaz 100% lenne a crossing-overek átlagos gyakorisága, 1 centimorgan (cM) távolság pedig 1% átlagos gyakoriságnak felel meg. A genetikai térképeken a távolságokat általában cM egységekre vonatkoztatják (1cM = a térkép egység). A M, cM mérőszám tehát nem egy metrikus, hanem egy statisztikus/valószínűségi dimenzió. A crossing-overek átlagos gyakorisága (tehát a M, cM érték) az elmélet szerint egyenesen arányos az a és b pontok/gének tényleges fizikai távolságával, azaz például kétszer nagyobb cM érték d-re kétszer akkora tényleges fizikai távolságot jelent. Ahogy fentebb említettük, a tesztelő keresztezésekben a crossing-overeket magukat nem látjuk, csak következményeiket észleljük, a rekombinánsokat. R, a rekombináció gyakorisága kísérletesen meghatározható (teszt-cross) és ebből d, a távolsággal egyenes arányban álló cM mérőszám, az átlagos crossing-over gyakorisági érték, a térképezési függvény alapján már kiszámolható. A genetika történetének leghíresebb kísérletei közé tartoznak azok, amelyek bizonyították, hogy a gének sorrendje a rekombináció alapján szerkesztett géntérképen megegyezik a géneknek (a térkép pontjainak) a kromoszómán, illetve a DNS mentén elfoglalt helyével. Azaz a két sorrend, a géntérképi és a valóságos, párhuzamba állítható, kolineáris. A géntérkép a maga szintjén helyesen tükrözi a valóságot, noha magát a DNS-szekvenciát nem láttatja. A molekuláris biológiai módszerek fejlődése azonban lehetővé tette, hogy képet kapjunk a cM távolságok valós fizikai méretéről. Ez a méret fajonként változik, és alapvetően a crossing-over mechanizmusnak a vizsgált élőlényre jellemző hevességétől függ. Az emlősök esetén 1 cM géntérképi távolság mintegy 1 millió bázispár hosszú DNS-szakasz (1 Mbp) fizikai távolságának felel meg a kromoszóma mentén (Drosophilában, vagy a C. elegans fonálféregben 20-50-szer kisebb, E.coli baktériumban, lambda-fágban még kisebb, 1000 bp körüli az értéke). Megjegyzendő, hogy a géntérképezési függvényben az úgynevezett idealizált „törés és újraegyesülés” crossing-over mechanizmussal számolunk (azaz: „a crossing-over pontszerű, véletlen, autonom esemény, a kromoszóma mentén bárhol azonos valószínűséggel léphet fel”), ami nagyon rövid DNS-szakaszokon már torzul (pl. a „génkonverzió”, a „crossing-over interferencia”, a rekombinációs „forró és hideg” pontok által), de nagyobb fizikai távolságok esetén (pl. 100 ezer bp), amelyek kicsiny töredékei egy emlősgenom teljes, 3 milliárd bázispárnyi (3GBp) hosszának, már közelít a valós crossing-over viselkedése az idealizálthoz. A lokális egyenetlenségek kioltják egymást. Az sem ritka, hogy a rekombináció hevesebb a női egyedekben, mint a hímekben, de a keresztezések megválasztásával kiküszöbölhető ez a hatás is. Az összehasonlító géntérképezés elve: minél közelebbi evolúciós rokonságban áll két faj annál jobban hasonlít géntérképük, nagy szakaszokon a gének sorrendje megegyezik, szinténikus. Ha a két faj géntérképét párhuzamba állítjuk és közülük egyikük genomszekvenciáját is ismerjük, a másik faj genomszekvencia-halmaza is nagyobb, szinténikus szakaszokba, s ezeken belül sorrendekbe rendezhető. Esetünkben rendelkezésünkre állt a gímszarvas közepes sűrűségű géntérképe, amely több mint 600 jól vizsgálható térképpontra, DNS-markerre épült. A térképpontok átlagos távolsága 5,4 cM volt. Feltételezhető volt, hogy ezek a térképpontok/DNS-markerek megtalálhatók a szarvasmarha géntérképén is, és nagy szakaszokon szinténikusak is, mivel szarvasmarha és a gímszarvas evolúciósan viszonylag közeli rokonok. A szarvasmarha részletes géntérképét és teljes genomja szekvenciáját a közelmúltban meghatározták. A kettős referencia így állt össze: ismert géntérkép a gímszarvas oldalán, és ismert géntérkép és a genom DNS-szekvenciája a szarvasmarha oldalán.
A milu, fajhibridek és visszakeresztezés
Külön ki kell térni arra, hogy miként készülhetett rekombináció alapú géntérkép a gímszarvasról, mert nem szokványos. A gímszarvasnak nincsenek különböző beltenyésztett fajtái, mint a háziállatoknak (pl. racka, cigálya, merino juhok stb.). A beltenyésztett vonalakban, fajtákban a beltenyésztés és a szelekció következtében a genom más-más részein homozigóta szakaszok rögzülnek, ami forrása a fajták genetikai különbségeinek is. A különböző fajták keresztezése megteremti a géntérképezés lehetőségét is. Egy másik eljárás a családfák, 3 generációs családok elemzésén alapul, amelyet pl. az első részletesebb emberi géntérkép meghatározásához használtak, s a szarvasmarháét is hasonlóan finomították. A szarvasok esetében más utat lehetett csak választani; az úgynevezett interspecifikus back-cross (fajhibrid visszakeresztezés) módszert, amikor a közeli fajok hibridjeit visszakeresztezik az egyik kiinduló fajjal. Az utódokban kimutathatók rekombinációs történések, ami lehetőséget ad géntérkép szerkesztésére. A közeli rokon fajokban ugyanis a génsorrendek a genomok nagy szakaszain, akár egész kromoszómákon, jó eséllyel megegyezhetnek (szinténikusak), de nagyszámú különbség is felhalmozódhatott a genomjukban azóta, hogy elváltak a közös ősüktől. Ezek a különbségek a mai DNS-diagnosztikai eljárásokkal viszonylag egyszerűen kimutathatók (pl. a mikroszatelliták: rövid, ismétlődő DNS-szakaszok variációi), felhasználhatók rekombinációs gyakoriságok meghatározásához és a géntérképezéshez. A gímszarvashoz evolúciósan közel álló faj a kínai császári kertek egykori szent szarvasa, a milu, más néven Dávid-szarvas (első leírója, Dávid atya jezsuita szerzetes után kapta a nevét: Elaphurus davidianus). A gímszarvas és a Dávid-szarvas keresztezhető, a fajhibridek életerősek és mindkét nemük termékeny. Ez szerencsés körülmény a géntérképezés szempontjából. Az emlős fajhibridek hímjei ugyanis (XY, a „heterogamétás szex”) általában sterilek, a női egyedek (XX, a „homogamétás szex”) ugyanakkor termékenyek. Ez az úgynevezett Haldane-szabály (látványos példa a szabályra az oroszlán és a tigris kétféle fiú hibridje, az oroszlán apa tigris anya hatalmas liger fia, és reciproka, a tigris apa oroszlán anya tigon fia, mindketten sterilek, míg lány testvéreik termékenyek). Mivel az általános Haldane-szabálytól eltérően a gímszarvas-milu hibrid hímje is fertilis, spermája alkalmas volt nagyszámú, a farmokon nevelt gímszarvas ünő mesterséges megtermékenyítésére. Teljesült a fajhibrid visszakeresztezése a kiinduló fajok egyikével! A mesterséges megtermékenyítésből született „back-cross” szarvasborjak (F2) nemzedéke jelentette a térképezésre alkalmas populációt. Több mint 700 borjútól vettek DNS-mintát és határozták meg a mikroszatellita/DNS-marker variációk között a rekombinációk gyakoriságait, ebből cM távolságokat számoltak – így szerkesztették meg a gímszarvas autoszómák géntérképét. Az X kromoszóma térképezéséhez egy további keresztezésre van szükség. Az F2 back-cross nemzedék ünőit kell keresztezni gímszarvashímekkel. Ezen ünők egyik X kromoszómája gím, a másik Dávid-szarvas eredetű a criss-cross öröklés miatt (a fiú az X kromoszómáját az anyjától kapja és a lányának adja tovább), ami lehetőséget ad a rekombináns X kromoszómák keletkezésére. Az F2 ünők X kromoszómáikat a fiaiknak (F3!) adják tovább. Az F3 fiúk X kromoszómáiból meghatározható az X kromoszóma géntérképe. Nagyszerű genetikai munka volt, amelyet John Slate és Diana Hill genetikus professzorok vezettek Új-Zélandon az 1990-es években. A géntérkép 621 géntérképi pontból épült, teljes hossza 2532 cM, amely a 33 autoszómának és az X és Y nemi kromoszómáknak megfelelően 35 kapcsolási csoportba rendeződik. A gímszarvas géntérképe kétszeresen is őrzi a híres brit genetikus Haldane (1892-1964) emlékét, a Haldane genetikai térképezési függvényben és a fajhibridek Haldane-szabályában.
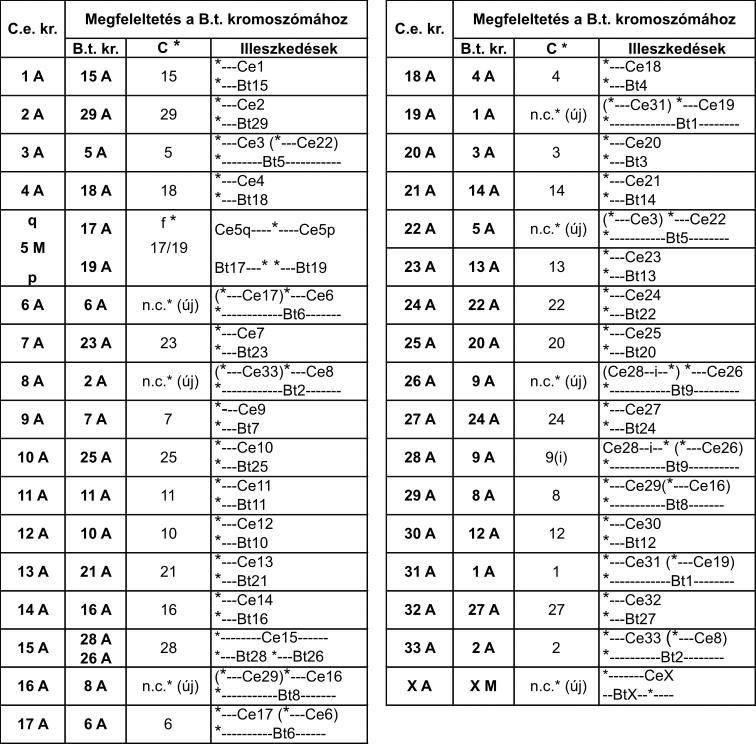
DNS-szekvenciától a gímszarvas-kromoszómákig
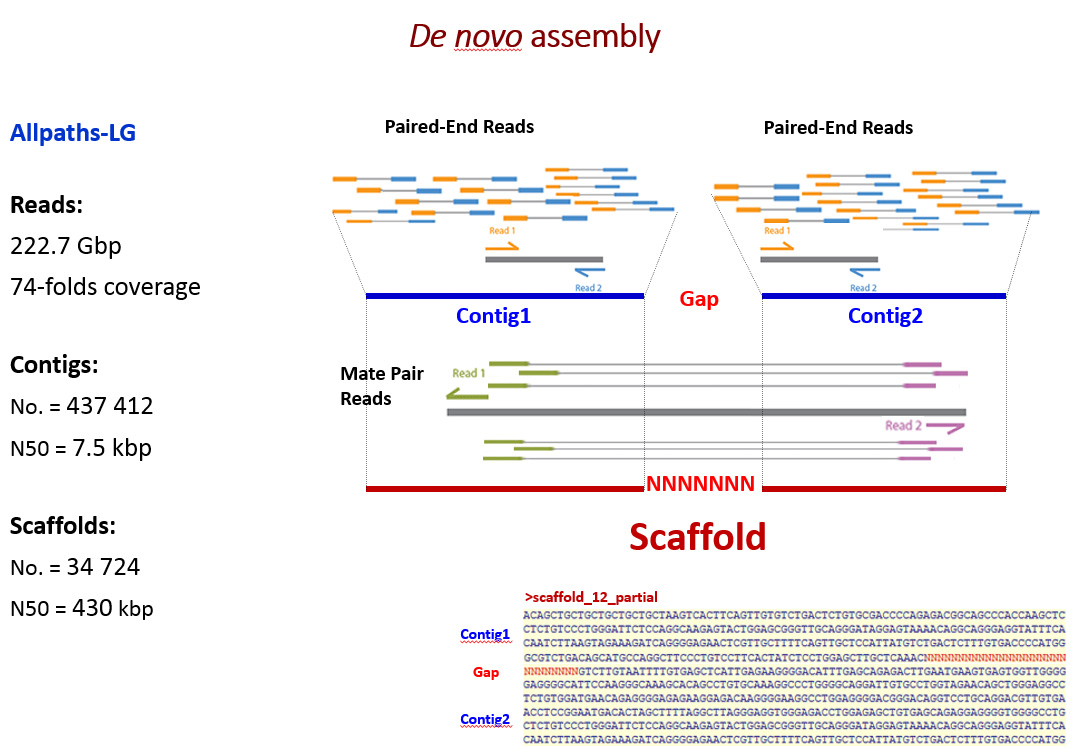
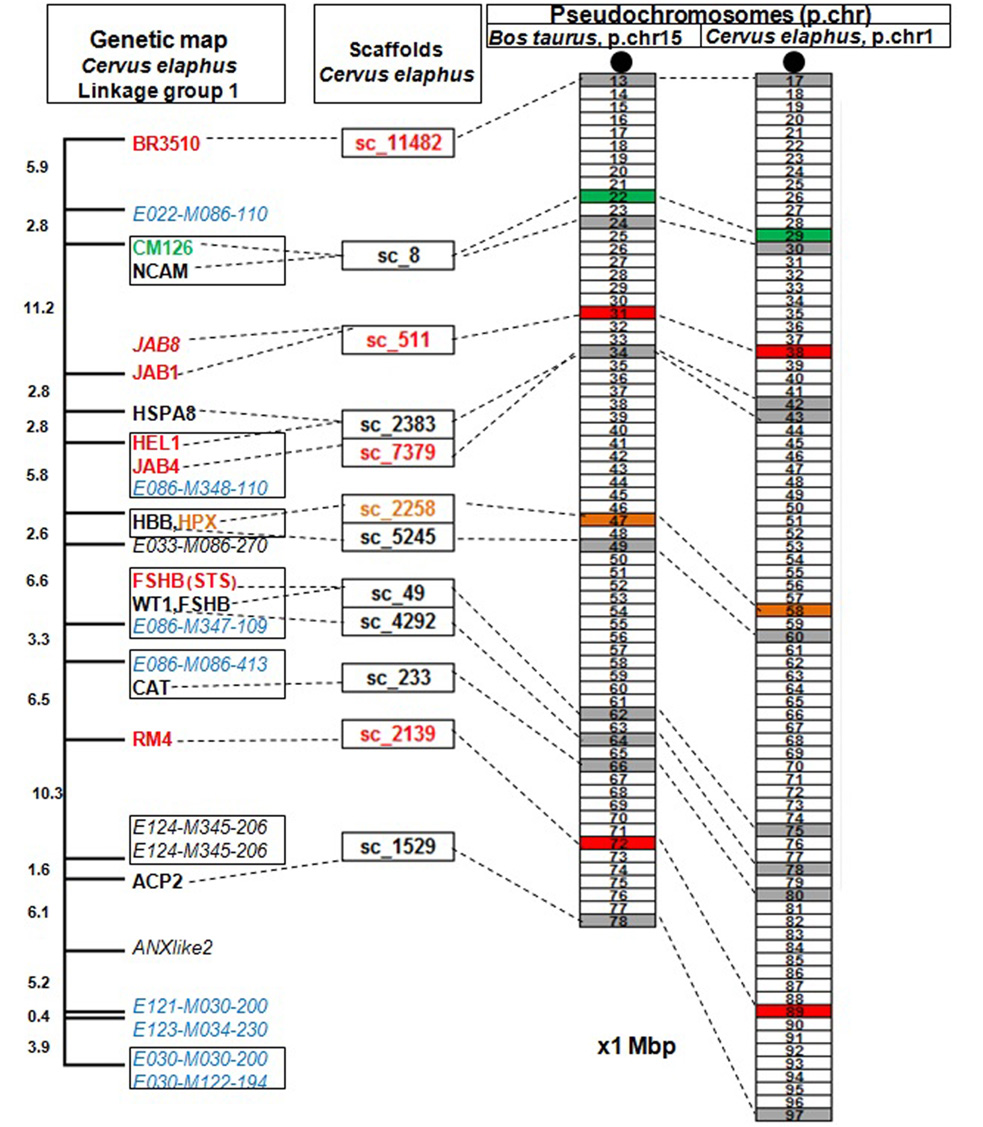
Az első lépés a DNS-mintavétel volt. Egy csodálatosan szép, kapitális szarvasbika 10 milliliternyi vérmintájából vontuk ki a DNS-t 2011-ben. A szarvasbika (Crot 3016, a cikk nyitó fényképének közepén, 7 éves korában 2010-ben) a Kaposvári Egyetem Vadgazdálkodási központja telepén, Bőszénfán élt, természetközeli körülmények között. Ezután hoztuk létre a DNS-könyvtárakat. Az izolált DNS-t először darabokra kell törni. A darabokból „DNS-könyvtárak” készülnek. A CerELa1.0 esetében kétfajta „DNS-könyvtár” készült, az egyik átlagosan 500 bp hosszú darabok halmaza volt (one paired library), a másik hosszabb daraboké (1,2 Kbp, mate pair library). A harmadik lépés a DNS-szekvenálás. A legmodernebb új generációs DNS-szekvenálási eljárások (NGS, New Generation Sequencing) egyikével (ILLUMINA Technology) meghatároztuk a DNS-könyvtárakban található valamennyi DNS-darab szekvenciájából a végeket (a bázisok sorrendjét) 101 bázispárnyi hosszon. Ezeket a szekvenciákat „read”-nek nevezik. A read-szekvenciák halmaza 74-szer több volt, mint az emlősgenom 3 milliárd bázispárnyi hossza, (3Gbp), azaz a genom minden pontját átlagosan 74-szeresen fedte le a meghatározott DNS-szekvenciák összessége (224 Gbp). Ezután következett a szekvenciaelemek építése („read és scaffold”). A hatalmas DNS-szekvencia-halmazt tovább rendeztük egy programcsomag segítségével (ALLPATH-LG). A read-szekvenciákat átfedéseik alapján folytonosan átfedő szakaszokba kontigokká („contig, continuus” DNS szekvencia) építettük, majd a hosszabb, mate pair könyvtári elemek végszekvenciáinak ismeretében mint valami ácskapoccsal, a kontigokat nagyobb egységekbe, scaffoldokba (szekvenciavázakba) lehetett fűzni. A scaffoldok (26 000) mérete széles tartományban szóródott (néhány 1000-től több millió bp-ig). Ezután történt a térképpont-scaffoldok (MapMarker, MMSc Scaffold) kihalászása. A gímszarvas genetikai térképe többségében olyan térképpontokat tartalmazott, amelyeket DNS-szekvenciák definiáltak (pl. mikroszatelliták, valós gének szekvenciai stb.). Ezeket a DNS-szekvenciákat mintegy „horgászcsaliként” lehet használni ahhoz, hogy a scaffoldok halmazából, a megfelelő számítógépes programmal „horogra akasszuk” azokat, amelyek ezeket a csaliszekvenciákat tartalmazzák (MMSc lett a nevük). További bioinformatikai lépésekkel meghatároztuk az MMSc-kben rejlő géneket. Ezzel a lépéssel a gímszarvas géntérkép térképpontjai gazdagodtak, a valós DNS és genomi környezetébe kerültek. Fontos lépés volt ezután a kolinearitás ellenőrzése. Az MMSc-ket visszapróbáltuk, visszaillesztettük a szarvasmarhagenom szekvenciájára (Btau_5.0.1. genom program). Ez a lépés egyben a gímszarvas-géntérkép és a szarvasmarhagenom kolinearitásának próbája is. Két fontos megállapítást lehetett tenni: egyrészt a szarvasmarhagenomon az MMSc-k által lefedett területeken kivétel nélkül, megtalálhatók voltak mindazon szarvas gének szarvasmarha megfelelői (ortológok), amelyeket az MMSc-k tartalmaztak, és ezek sorrendje is egyező volt. Tehát az MMSc-ken belüli szinten szinténikus (egyben kolineáris) a szarvasmarha- és a szarvasgenom. Másrészt az MMSc-k sorrendje 19 szarvas autoszóma és az X és Y kromoszómák esetén tökéletesen kolineárisak a szarvasmarha-kromoszómákkal. A további esetekben is érvényesült a kolinearitás, ha figyelembe vettük a szarvas és a szarvasmarha evolúciós útján történt kromoszómaátrendeződéseket („kariológiai evolúció”: az ősi kromoszómák kettéhasadása, két ősi fúziója eggyé, transzlokációk, inverziók). Összefoglalva: az MMSc-k sorrendje és az MMSc-ken belüli gének sorrendje egyaránt megfeleltethető volt a szarvas és a szarvasmarha genomban, ezeken a szinteken érvényesült a kolinearitás. Ezután határoztuk meg a referenciagéneket hordozó scaffoldokat (RGSc-ket). A szarvasmarha genetikai adatbázisában külön kiemelésben listázták a „valamilyen szempontból” fontos géneket (referenciagének). A mi előzetes kísérleteink tapasztalata az volt (több mint 100 szarvasgént klónoztunk előzetesen), hogy a két faj ortológ génjeinek szekvenciája átlagosan legalább 95%-ban megegyezik, ami kézenfekvővé tette, hogy a szarvasmarha referenciagének szekvenciáival „horogra akaszthatók” az ortológ szarvasgéneket tartalmazó scaffoldok, az RGSc-k. Több mint 6000 RGSc genetikai tartalmát lehetett így meghatározni, ami nagyon hasonló eredményt adott az MMSck-nél tapasztaltakkal. Az RGSc-kben a két faj ortológ génjei voltak azonos sorrendben. Az RGSc-ket az MMSc-k által közrefogott területekbe a szarvasmarhagenom sorrendjét követve töltöttük be. Bíztunk abban, hogy a kolinearitás zömében érvényesülni fog az RGSc-k sorrendjére is. (Reményünk jogossága bebizonyosodott, lásd alább) Az MMSc-k és RGSc-k közötti hézagokat eztán feltöltöttük IRGSc-kel (inter referencia gén scaffoldok). A maradék scaffoldok halmazából kihalásztuk mindazokat, amelyekben az emlősökre jellemző gének vagy gének egy részlete azonosítható volt bioinformatikai eszközökkel (ezek az IRGSc-k). Az IRGSc-ket elhelyeztük az MMSc-k és RGSc-k közötti hézagokba. Néhány bizonytalan esetben a sorrendek meghatározásához kapaszkodót adott több más emlősgenom programja is, pl. a birkáé. Végső lépésként – itt nem részletezett komplex úton – meghatároztuk a centromeronok helyét. A centromeronok biztosítják sejtosztódáskor a kromoszómák precíz vándorlását az utódsejtekbe. Hiányuk összeegyeztethetetlen az élettel. Feltárult, hogy a szarvas és szarvasmarha evolúciójakor mely kromoszómákon egyezik, hol keletkezett új, másutt hol veszett el az ősi Pecora kromoszómától örökölt centromeron. Eme lépések és a gondos ellenőrzések után 26 108 szekvenciaelemet (döntően scaffoldokat és néhány önálló contigot), több mint 20 ezer gént (a kérődzők, emlősök ismert génjeinek 90%-át) rendeztünk el a szarvas géntérképéhez és kromoszómáihoz igazítva. A gének kromoszómák szerinti helye, valamint sorrendje a scaffoldokon belül bizonyosan a szarvas genomjának felel meg.
Független visszaigazolás
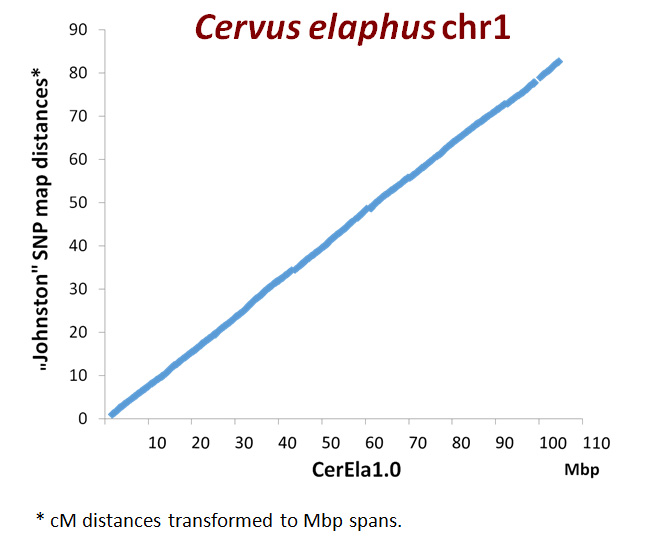
Miközben a CerEla1.0 nyomdában volt, elérhető lett egy skóciai–új-zélandi kutatók által készített nagyon sűrű, 38 000 SNP-pont rekombinációjából szerkesztett szarvas- géntérkép, amelyen 100-200 ezer bázispár választja el a térkép pontjait. Ez mintegy 50-szeresen nagyobb térképsűrűség annál, mint amit a CerEla1.0 összeállításánál alkalmazni tudtunk. Nagyon jó felbontásnak számít. Lehetőséget adott arra, hogy kromoszómánként átlagosan több, mint 1000 térképponthoz lehessen igazítani a CerEla1.0-t . Bana Nóra és Nyíri Anna doktoranduszok az MGG-ben megjelent publikáció megosztott első szerzői visszafelé is elvégezték a kolinearitás tesztjét. A vizsgálat függetlenül igazolta, hogy a CerEla1.0 hitelesen ésnagy pontossággal írja le a gímszarvasgenomot. A skót–új-zélandi kutatók szerkesztette géntérképet „ráfektettük” a gímszarvasgenom DNS-szekvenciájára, a CerEla1.0 szekvenciára. Az új, nagy sűrűségű géntérkép pontjai, néhány apró kivételt nem számítva, azonos sorrendben helyezkedtek el a CerEla1.0 szekvencia mentén is.
Annotálás és elhelyezés adatbázisokban
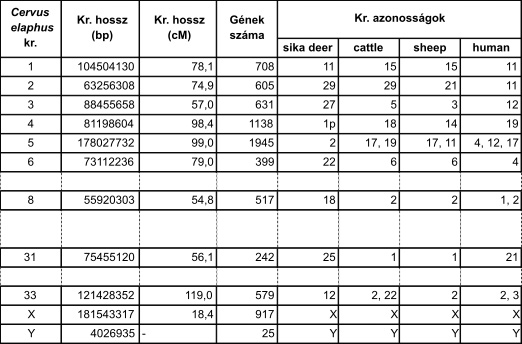
A CerELa1.0 genom elérhető és letölthető az NCBI adatbázisból (NCBI, MKHE00000000 azonosítóval), továbbá az MBKNAIK honlapról is (elérési útvonal: http://http.//emboss.abc.hu/wonderdeer/JBrowse) és elérhető a Kaposvári Egyetem és az ELTE Genetikai Tanszékén (https://genetics.elte.hu/index.php/hu/) is. A CerEla1.0-ról beszámoló MGG-közlemény elérhető a folyóirat honlapján, de érdeklődőknek a jelen cikk szerzője interneten is elküldi. A CerEla1.0 adatbázisokban „zoomolva” a genom egyre mélyebb rétegei ismerhetők meg, egészen le az alapokig, a DNS-szekvenciáig: letölthető a genom teljes szekvenciája, egyes kromoszómáké. Az 1. kromoszóma pl. 104.5 millió bázispár hosszú DNS-t tartalmaz és 708 gént hordoz. A leghosszabb autoszóma az 5., amely 178 millió 30 ezer bázispár és 1945 gént kódol. A legrövidebb 55 millió 920 ezer bázispárral a 8., a legkevesebb gént a 31. hordozza, 242-t. Az ivari kromoszómákról: valamennyi kromoszóma között az X a leghosszabb, az Y a legrövidebb. Tetszés szerint, bázispár pontossággal kijelölhető bármely kromoszóma egy kisebb részlete. Kiolvasható a gének leolvasási iránya, az exon-intron szerkezet, a gén funkciója és megfelelőik (az „ortológok”) más fajokban, a gén által kódolt fehérje aminosavsorrendje, a gén fajon belül megismert változatai, az allélok. Megjeleníthetők „madártávlati nézetek”, pl. az ismétlődő szekvenciák és a különböző gének (fehérjét kódol, tRNS-t, riboszómaRNS-t, kis RNS-t, transzpozont stb.) sűrűségéről, a géntérképezés szempontjából fontos vonatkoztatási pontokról. Kikereshető, hogy a genomját adó szarvasbika a genom mely pontjain heterozigóta, és böngészhető a teljes genetikai változatosság is a genom 32 000 pontján egy 4500 egyedet számláló természetes gímszarvas-populációban.
Miért éppen a gímszarvas?
A szarvasok genetikai vizsgálatának gondolata már négy évtizede felmerült Szegeden az egyetem (JATE) Genetikai Tanszékén és az SZBK Genetikai Intézetében egy gemenci kirándulás után. Az agancs, ez a komplex csontos szerv, a „macsó” dominancia extravagáns megjelenítője („fenotípus”), évről évre a tél végén lehull, majd néhány őssejtből regenerálódik. Három hónap alatt akár 10-15 kg súlyú trófeává fejlődik, ami összemérhető a szarvasbika vázcsontozatának negyedével-harmadával. Az agancs fejlődése a legintenzívebb szövetgyarapodás az állatvilágban, a rosszindulatú tumoroknál is sokkal hevesebben, naponta akár 20 dkg-mal gyarapodik, és ágvégein 1-2 cm-t nő. Arra jutottunk, hogy a táplálkozás önmagában nem biztosíthatja az agancs csonthoz szükséges kálcium mennyiségét, s a kiegészítés csak a vázcsontozat bomlás árán halmozódhat át, amelyet Gödöllőn, a Biotechnológiai Központban igazoltunk. Azaz a szarvasbika csontritkulást szenved egy ideig, de az agancs elkészültével, július-augusztus során helyre állítja csont sűrűségét, „meggyógyítja magát”. Erre az ember szervezete nem képes, noha kimutattuk, a SOTE oszteoporózis-kutatóival együttműködve, hogy a csontritkulásban szerepet játszó gének és genetikai útvonalak nagyon hasonlóak emberben és szarvasban. Évszázados, talán több évezredes tapasztalat, hogy a szarvas jól tenyészthető félig vad, természetközeli tartásban. A Kaposvári Egyetem az 1970-es években egy kísérleti szarvasfarmot épített Bőszénfán, gyönyörű környezetben, folyamatosan fejlesztve a legmodernebb eljárásokkal (pl. mestersésges megtermékenyítés, embriótranszfer, CT-vizsgálatok, származáskövetés stb.). Szarvasembriókat szállítottak beültetésre pl. Új-Zélandra („szarvas-lombikbébi”- program), ahol a „farm szarvas” tenyésztés nemzeti iparággá fejlődött. Mindeközben idehaza a molekuláris genetika új intézménnyel bővült az 1980-as évek legvégén, Gödöllőn: a Mezőgazdasági Biotechnológiai Központtal (MBK), amely infrastruktúrájában, műszer- és informatikai felszereltségével a legfejlettebb országok legjobb laboratóriumaival is állta az összehasonlítást. Az 1990-es évek végére az MBK-ban fejlődött ki a szarvasok molekuláris genetikáját kutató laboratórium és hálózat. Találkozott itt a géntérképezéssel, fágokkal, baktériumokkal foglalkozó fiatal molekuláris genetikus az emlősök klónozásával, vírusaival foglalkozókkal, vegyészekkel, akik DNS-t és peptideket tudtak szintetizálni és szekvenálni. A munkatársak, diplomamunkás MSc-sek, doktoranduszok már a Szegeden egykor kialakult klasszikus és molekuláris (integrált) genetikakurzusokon edződtek. Csak idő kérdése volt Gödöllő, Kaposvár, az ELTE és a SOTE doktori iskoláinak összefogása. A szarvasok közös nevezőt adtak a CerEla 1.0 felé vezető közel két évtizedes úton. A CerEla1.0 folyamatosan bővíthető az új ismeretek függvényében, gazdag forrása máris populációgenetikai elemzéseknek (pl. szarvasok egyedi azonosítása, illetve szarvaspopulációk eredetének megállapítása), leszármazási és eredet vizsgálatoknak, forenzikus, bűnügyi, vadorzási, régészeti, természetvédelmi és vadgazdálkodási felhasználásoknak. Meghatározhatók voltak pl. a Kárpát-medencei gímszarvasok anyai és apai leszármazási vonalai (haplotípusok, „vérvonalak”), s az is, hogy milyen útvonalakon népesítették be a szarvasok a Kárpát-medencét a jégkorszak után. Több ezer éves szarvascsont-maradványok DNS vizsgálata („archeogenetika”) mutatta meg, hogy merre élnek a mai leszármazottak. DNS vizsgálatok segítettek bírósági esetekben, vadorzók elítéléséhez. A CerEla1.0 teljes genomasszociációs vizsgálatokra (GWAS, Genome Wide Assotiation Studies) ad lehetőséget. Egy ilyen látványos GWAS-vizsgálat lehet a jövőben például az aranyérmes trófeák titkának megfejtése is, azaz hogy milyen mértékben öröklődik a kapitális agancs (mekkora a heritabilitás), mely gének mely változatai és működési hálózatuk van a háttérben (a „hajlamosító” gének hálózata). Az utóbbi években a CerEla1.0 számos természetvédelmi célú genomkutatás sikeres elindításának jelentett bátorítást a „gímszarvasgenomon” felnőtt fiatal kutatóknak, Stéger Viktornak , Frank Krisztiánnak és doktoranduszcsapatuknak. Ilyen például a nagy ragadozók (medve, farkas, hiúz) magyarországi előfordulásának és vándorlásának igazolása, vagy a vaddisznóállományok nyomon követése genomdiagnosztikával. A pannon méh genomjának feltárása és a DNS-vizsgálat méhészeti alkalmazása is innen nőtt ki. A CerEla1.0 nagyban megkönnyíti DNS-diagnosztikai eljárások és genomvizsgálatok kidolgozását más vadfajokra, pl. dámvadra, őzre, muflonra. A CerEla1.0 hasznosult már eddig is az emberi oszteoporózis kutatásában, és hasznosulhat a jövőben a fejlődés és orvosi biológia területein (pl. a szerv fejlődés, a szerv és szövet regeneráció, robusztus szövet gyarapodás és tumor biológiai kutatásokban).




RÖVIDÍTÉSEK
- bp: DNS-bázispár, Kbp: kilo bp, 1000 bp, Mbp: mega bp, 1 millió bp , Gbp: giga bp, 1 milliárd bp,
- cM: 1% a crossing overek átlagos előfordulása két pont/gén között a géntérképen, a térképegység. A genetikai térképezési függvény szerint a cM érték egyenesen arányos a két térkép pont valós fizikai távolságával.
- Ortológ gének: különböző fajokban azonos funkciót ellátó gének, DNS szekvenciájukban hasonlítanak.
- Szintenikus gének: egyazon kromoszómán/kapcsolási csoportban helyezkednek el.
- Homológ kromoszómák: az egymásnak megfeleltethető kromoszómák.
IRODALOM
[1] Bana Á.N. et al.(2018) The red deer Cervus elaphus genome CerEla1.0: sequencing, annotating, genes, and chromosomes. Mol Genet Genomics 293:665-684,
[2] Slate J. et al. (2002) A deer (subfamily Cervinae) genetic linkage map and the evolution of ruminant genomes. Genetics 160: 1587-1597,
[3] Johnston S.E. et al.(2017) A high-density linkage map reveals sexual dimorphism in recombination landscapes in red deer (Cervus elaphus). G3 7: 2859-2870,
[4] Btau_5.0.1 – Genome – Assembly – NCBI https://www.ncbi.nlm.nih.gov/assembly/585021 Cattle. Bos taurus (cattle) genome assembly Btau_5.0.1 from Cattle Genome Sequencing International Consortium [GCA_000003205.6 GCF_000003205.7]
A cikk a Természet Világa 2018. szeptemberi (149. évf. 9. sz.) számában jelent meg.
