KIRÁLY PÉTER ATTILA
Az öröklődő leukémia
A
tévében és a hirdetésekben látható kihullott hajú kisgyermekek, steril szobák
és meggyötört arcú szülők képe már beleégett emlékeinkbe a gyermekkori leukémia
szó hallatán. Habár ezek a leukémiák még ma sem gyógyíthatók 100%-ban, azt
mondhatjuk, hogy a széleskörű kutatásnak és a gyógyszerfejlesztésnek
köszönhetően az onkológia egyik legnagyobb sikertörténete.
Érdekes
módon, a leukémiáknak, és különösen a gyermekkori, valamint fiatalkori
leukémiáknak léteznek olyan megjelenési formái, amelyek ugyanabban a családban
előfordulva akár az összes gyermeket érinthetik. Ezek az úgynevezett örökletes
leukémia-szindrómák (familiáris leukémia). Az ilyen leukémiák kialakulása
tisztán genetikai okokra vezethető vissza – tehát nincsenek környezeti
hajlamosító tényezők –, és egy meghatározott gén mutációja vagy mutációi
kapcsán alakulnak ki. Írásomban ezeket a leukémiákat mutatom be részletesebben,
mert az elmúlt évek kutatásai alapján kiderült, hogy a kórképek familiáris
megjelenési formája korántsem ritka jelenség.
Örökítő
anyagunk, a DNS sejtjeink belsejében a sejtmagba zárva található, és
úgynevezett hisztonfehérjékre tekeredve extrém tömör és kompakt formába
rendeződik. Ha egyetlen sejt DNS-tartalmát „kitekernénk”, az mintegy két méter
hosszúságú lenne. A DNS-molekula azon szakaszait, melyek fehérjévé „íródnak”
át, géneknek nevezzük, ám ezek érdekes módon a teljes genetikai állománynak
csupán 1–2%-át teszik ki. A DNS építőkövei – az adenin, a citozin, a guanin és
a timin – nukleotidmolekulák, melyek egymás utáni kombinációja alkotja teljes
genetikai állományunkat. A fehérjék a sejtek valamennyi feladatának ellátásáért
felelősek, ide tartoznak a sejtek szerkezeti, metabolikus és hírvivő funkciói,
ám természetesen a sejtek osztódása, szaporodása is. Ha a kódoló génszakaszban
a nukleotidok sorrendje megváltozik (megrövidül/egy vagy több nukleotid kiesik,
vagy kicserélődik más nukleotidra) és ez károsítja az adott fehérje működését,
mutációról beszélünk. Könnyű ezek után elképzelni, hogy ha egy mutáció olyan
génben jelenik meg, amely a fehérvérsejtek életében fontos szerepet játszik, az
a leukémia kialakulásának kockázatával jár. Ha tudjuk, hogy a nem örökletes
daganatok kialakulásában – így a nem családi halmozódású leukémiákban is –
szintén mutációk játszanak szerepet, akkor mi a különbség közöttük? Ha egy
mutáció csak a tumorsejtekben van jelen, tehát abban az alapszövetben
(esetünkben a csontvelőben) jelenik meg, amelyből a daganat kialakul,
úgynevezett szomatikus mutációról beszélünk. Egy adott daganatsejtben akár több
tucat ilyen mutáció is lehet, így egy daganat – amelyet sok egymástól
genetikailag eltérő tumorsejt alkot – akár több száz különböző mutációt is
hordozhat. Ha egy mutáció a szervezetet felépítő összes sejtben jelen van
(tehát a nem daganatos sejtekben is), úgynevezett csíravonalbeli vagy öröklődő
mutációról beszélünk. Ezek a csíravonalbeli mutációk valamelyik szülőtől
származnak, és a genetika alapszabályai alapján 50% eséllyel öröklődnek
generációról generációra. A familiáris leukémiákban olyan génekben lehet
örökletes mutációt kimutatni, amelyek a vérsejtek érésében vagy működésének
szabályozásában játszanak szerepet.
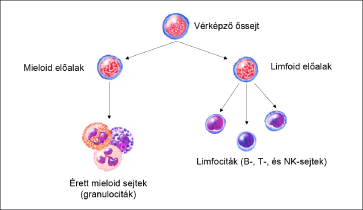
A
vérképzés rendszerének sematikus ábrázolása
A
leukémiák típusai és főbb jellemzői
A
másik sejtvonalból kiinduló daganatok a mieloid leukémiák, melyek döntően a
felnőttkor, azon belül is az idősebb életkor betegségei, de bármilyen
életkorban előfordulhatnak szindrómák kapcsán, vagy családi halmozódásban.
A
heveny leukémiák, nevükből is adódóan, többnyire súlyos és gyors lefolyású
megbetegedések. A leukémia szó szerinti fordításban kóros fehérvérűséget
jelent, aminek során kóros fehérvérsejt-alakok jelennek meg a csontvelőben és a
perifériás vérben. A leukémiás sejtek kiszorítják a csontvelőből az egészséges
vérképzést, aminek eredményeképpen eltűnnek a funkcióképes immunsejtek,
vörösvértestek és vérlemezkék a keringésből. Gondoljuk végig a súlyos
következményeket. Immunsejtek hiányában a legbanálisabb kórokozó is komoly
veszélyforrássá válik, szinte leküzdhetetlen fertőzések alakulnak ki.
Vörösvértestek hiányában súlyos fáradékonyság, letargia és elesettség alakul
ki. Vérlemezkék hiányában pedig az akár apró sérülések helyén is
csillapíthatatlan vérzések jelenhetnek meg, valamint előfordulhatnak spontán
bőrbevérzések (ez jellegzetes tünet, az orvosi terminológiában purpuráknak
hívjuk és megjelenésük esetén javasolt azonnal orvoshoz fordulni). Minden tünet
önmagában is kellő súlyosságú, együttesen azonban még ijesztőbb képet
kölcsönöznek.
A
gondozásba vett betegeken, így a gyermekeken is azonnal el kell végezni a
széleskörű laboratóriumi vizsgálatokat, rögzíteni kell a csontvelő mintavételét
és a teljes kórtörténetet (családi kórtörténetet is). A kórtörténet
rögzítésekor figyelni kell számos korábbi és a családban már előfordult
betegségekre (hematológiai és nem hematológiai betegségekre egyaránt). Az
örökletes leukémiák esetén ezek a jellegzetes tünetek – különleges fertőzések,
csontvelőkimerülés jelei, különböző súlyosságú vérlemezke eredetű véralvadási
zavarok – rendszerint fiatal életkorban jelentkeznek gyakran több, hasonlóan
fiatalkorú családtagnál. Sokáig úgy tartották, hogy ezen örökletes kórképek
inkább ritka, elszigetelt irodalmi esetek, és a gyermekkori mieloid leukémia
(bár hangsúlyozni kell, hogy a gyermekkori mieloid leukémiák alapvetően ritka
kórképek) inkább sporadikusan, véletlenszerűen fordul elő, genetikai kódunk
megismerésével azonban változott ez a kép.
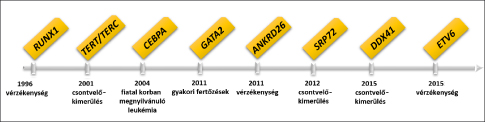
A
familiáris hematológiai kórképek hátterében álló főbb genetikai eltérések,
felfedezésük éve és a gén mutációja kapcsán kialakuló klinikai tünetcsoport
Technológiai
vívmányok a leukémia kutatásának szolgálatában
Tegyünk
itt egy újabb kitekintést a sejtbiológia, azon belül is a genetikai kód
megismerésének történetére, és technikai vívmányaira. Amikor 2003-ban a Humán
Genom Project (HUGO) vezetői bejelentették, hogy feltérképezték a teljes emberi
genomot (azaz mind a közel 3 milliárd nukleotid sorrendjét azonosították, mely
az emberi gének és nem kódoló régiók összességét alkotja), a technológiai
eljárások még lassúak és munkaigényesek voltak. A génszekvenálás klasszikus
módszerével a DNS-lánc felépülését mesterséges körülmények között idézték elő,
lépésenként megállítva a folyamatot, és meghatározva, milyen nukleotid épült
be. A HUGO segítségével ekkor még több milllió dollárból, és csak hónapok alatt
lehetett nagyobb mennyiségű genetikai információt megismerni. Az új évezred
azonban hatalmas technológiai robbanást hozott ezen a tudományterületen is. A
kémiai, elektronikai és informatikai módszereknek köszönhetően a génszekvenálás
folyamata a több hónapnyi időről mindössze néhány órás időtartamra csökkent, és
lehetőség van az így keletkező óriási adatmennyiség informatikai feldolgozására
is, hiszen ma már egy önálló tudományág képviseli ezt a területet: a
bioinformatika. Ezt a technikai fejlődést minden biológiai tudomány
hasznosítani tudta. Ennek köszönhetően számos faj genetikai kódját megismertük,
össze tudjuk hasonlítani a különböző fajok génállományát, és ami az
orvostudomány szempontjából nagyon fontos: nagy mennyiségű adatot ismerhettünk
meg a különböző betegségek genetikai hátteréről.
A
familiáris leukémiák megismerése és genetikai háttere
Fiatalkorban,
megelőző hematológiai betegségek nélkül jelenik meg az akut mileoid leukémia
(AML) a CEBPA gén mutációja miatt. A CEBPA gén fontos karmester a vérsejtek,
ezen belül a mieloid elemek érése és fejlődése során. Az ép CEBPA gén terméke
egy génátíródásért felelős faktor, mely a megfelelő időpontban be-, illetve
kikapcsolódva irányítja a sejtek érését és fejlődését. Mutációja ismert
sporadikus leukémiákban is, sőt ezekben az esetekben kifejezetten jó
betegségprognózissal társul, és jelen ismereteink szerint transzplantáció
nélkül gyógyítható. Családi halmozódás hátterében is leírták szerepét, ilyenkor
a csíravonalbeli gén érintett. A betegség egyetlen tünete (szinte 100%-os
valószínűséggel) a fiatalkorban kialakuló akut mieloid leukémia. A szokatlanul
fiatal életkorban jelentkező betegek esetén ésszerű a gén vizsgálatának
elvégzése a donorválasztás és a hordozók azonosítása szempontjából.

Familiáris
eset kivizsgálásának algoritmusa. A családfa felvétele után az érintett egyének
vérmintáiból fehérvérsejteket szeparálunk, ezekből DNS-t izolálunk és
meghatározzuk az egyén mutációs státuszát
Jellegzetes
és érdekes kórképek tartoznak a GATA2 gén csíravonalbeli mutációjával
jellemezhető betegségcsoportba. A GATA2 szintén szabályozó faktor a
vérképzésben, ám jellegzetesen két időpontban fontos a működése. A hemopoetikus
őssejt szintjén, ahol az önmegújító képességben játszik szerepet, valamint
később számos sejtféleség (limfoid és mieloid) végső érése kapcsán. Közös
jellemzője ezeknek a kórképeknek, hogy ugyanazon gént érintő mutáció esetén
mégis többféle betegség nyilvánul meg. Ezek javarészt immunológiai kórképek
(hiszen a fehérvérsejtek funkciói károsodnak), közös jellemzőjük pedig a magas
hajlam a leukémia kialakulására. A GATA2 gén mutációjánál a teljes élethossz
alatt mintegy 80%-os valószínűséggel alakul ki leukémia, döntően 35 év alatt.
A
leggyakrabban előforduló familiáris leukémia-szindróma, a familiáris vérlemezke
funkciózavarok talaján kialakuló mielodiszplázia (a fehérvérsejtek kóros alaki
és mennyiségi megjelenése) és akut mieloid leukémia. A klinikai kép ebben a
betegségcsoportban nagyon változatos. Egyes betegekben klinikailag teljesen tünetmentes
vérlemezkeszám-eltérések, esetleg enyhe, középsúlyos vérzészavarok alakulnak
ki, míg másoknál leukémia jelenik meg. A nagy heterogenitás okát intenzíven
kutatják. Kialakulásában valószínűleg a később megjelenő, ún. súlyosbító
mutációknak van szerepe. A kórkép hátterében a RUNX1 nevű vérsejtérést
szabályozó faktorban található csíravonalbeli mutációk állnak. Jól látható,
hogy ez már a harmadik olyan kórkép, melyet szabályozó gének zavara okoz. Ez
nem meglepő, hiszen könnyen elképzelhető, hogy csontvelőnkben, ahol a teljes
élet során napi több millió sejt képződik, egy szabályozó molekula kiesése
milyen károsodást képes előidézni.
A
másik ilyen nagy csoport a telomért érintő kórképek, melyek fiatalkori
csontvelőkimerüléssel, illetve számos egyéb klinikai megnyilvánulással
járhatnak. A telomérák szerepe a sejtbiológia egyik legfrappánsabb jelensége,
és szerepük szinte kézenfekvőnek tűnik ezekben a kórképekben. A kromoszómakarok
legvégén, mintegy sapkaszerűen ülnek a telomérrégiók, melyek a sejtek biológiai
órájaként működnek. Ugyanis minden osztódáskor rövidülnek, és egy kritikus
rövidséget elérve, a sejt nem osztódik többet, elhal. Hogy ez ne okozzon gondot
az olyan szövetekben, ahol az élet végéig folyamatos osztódásra van szükség
(például a csontvelőben), egy finoman szabályozott rendszer visszaépíti a kieső
szakaszokat. A familiáris csontvelőkimerülés kapcsán ezekben a rendszerekben
találunk csíravonalbeli mutációkat, aminek következtében károsodott az aktívan
osztódó szövetekben a telomérákat védő mechanizmus. Ezekben a betegekben
gyakran a hematológiai megbetegedést megelőzően több fejlődési rendellenesség
is felhívhatja a figyelmet a betegségre, azonban az sem ritka, hogy az első
tünet a gyermekkori vagy fiatal felnőttkori csontvelőkimerülés. Ilyenkor a
vérsejtalakok eltűnnek a perifériáról és a sejtek hiánya számos klinikai
tünetet okozhat. A vérképzősejtek azonban folyamatosan túlhajtott állapotban
vannak a hiányállapotok miatt, aminek kapcsán tovább sérülhet a génállományuk.
A
familiáris leukémiák kutatása és diagnosztikája Magyarországon
Aki
génjeiben hajlamot hordoz a leukémia kialakulására, de a vizsgálat idejében még
nem beteg, annál nyomon kell követni a változásokat, mintegy várva a betegség
első jeleinek megjelenésére a gyors beavatkozás érdekében (3. ábra). A hordozó
rokonok és érintettek esetében a családtervezés is fontos szempont a
továbbiakban. Kutatócsoportunk egyik fő érdeklődési területe az ilyen családok
összegyűjtése, és a hajlamosító genetikai tényezők feltárása, hozzájárulva
ezzel a betegségek biológiájának jobb megértéséhez. A közelmúltban több
családot is azonosítottunk, akiknél meghatároztuk a betegség hátterében álló
csíravonalbeli mutációkat, egy esetben pedig lehetőségünk volt a család
valamennyi tagjában szimultán elvégezni az összes kódoló génszakasz
vizsgálatát, hogy jobban megértsük, milyen további genetikai lépések vezettek
el a betegség kialakulásához. Munkacsoportunk ezen felül, hazánkban elsőként,
kidolgozott egy diagnosztikus eljárást, amely magában foglalja valamennyi
jelenleg ismert csíravonalbeli mutációs célpont vizsgálatát, hozzájárulva ezzel
e ritka és különleges betegcsoport pontosabb azonosításához és hatékonyabb
kezeléséhez.
A
Magyar Tudományos Akadémia Természettudományi Kutatóközpontja (MTA TTK) és a
Tudományos Ismeretterjesztő Társulat (TIT) közös ismeretterjesztő
cikkpályázatára beérkezett írás.
| Természet Világa, | 147. évfolyam, 11. szám,
2016. november
http//www.termvil.hu/ |