Genomika az orvosbiológiában
Napjaink kísérletes biológiája, az élõ rendszerek molekuláris mûködésének megismerése és alkalmazása óriási változáson megy át. Ennek legfontosabb mozgatóereje a genomikai megközelítés bevezetése. A genomika genom léptékû biológiát jelent, vagyis azt, hogy a vizsgálatok kiterjedhetnek akár az adott élõlény (például az ember) összes génjének DNS-szintû, illetve expressziós (mRNS és/vagy fehérje) analízisére. A teljes genom DNS-szintû megközelítését szerkezeti, az expressziós vizsgálatokat pedig funkcionális genomikának nevezzük.
A genomika abban segít bennünket, hogy a gének funkcióit megértsük, ami a modern molekuláris sejtbiológiában és a biológia szinte minden ágazatában korszakváltást hoz, gyakorlati szempontból pedig a biomedicinában és a mezõgazdaságban idéz elõ rendkívüli változásokat. Hangsúlyozni kell, hogy mai nézeteink, a gyakorlati felhasználás hatóköre és a kivitelezés logisztikája feltehetõen gyorsan módosulni fognak.
A genomika alkalmazását három, tudománytörténeti értelemben egyenként is jelentõs terület egyazon idõben született eredményei tették lehetõvé. Az elsõ lökést a genomprogramok adták. Mint tudott, 2001 elsõ hónapjaiban már a baktériumok, az élesztõ, a szõlõmuslica, a fonálféreg és a növények mellett az emberi genom, legújabban pedig az egér lényegében teljes genomiális géntérképe elkészült, a világháló adatbázisaiból lehívható és elemezhetõ. Bár a gének annotációja (azonosítása) még sok idõt vesz igénybe, ez a lexikális tudásanyag új genetikai információs minõség. Rendelkezünk ma már összehasonlító, illetve funkcionális szempontból csoportosított adatbázisokkal is. A legtöbb genomiális adat minden megkötöttség nélkül hozzáférhetõ, a szabadalom alá esõ gének elzártsága is (legalábbis emberben) idõleges. A második terület, amely lendületet adott a genomika alkalmazásának, a microarray (csip) módszer, aminek speciális jellegzetességei nagyságrendekkel emelik az egyidejûleg vizsgálható gének számát, s ami szerkezeti (nukleotidsorrend) és funkcionális (génkifejezõdés-mRNS) információk tömegét képes nyújtani. A harmadik tudományterület a bioinformatika. Ez az új biostatisztikai/biomatematikai megközelítés korrelációs és halmazelméleti eljárásokkal elemzi a genomiális/expressziós adatbankok és a géncsiptechnika által szolgáltatott adathalmazt, és biológiai következtetésekre alkalmas elemzést nyújt.
Rövid írásomban elsõsorban a microarray technológiáról, felhasználásáról és eddig felismert alkalmazási területeirõl, valamint a bioinformatikáról adok áttekintést.
A géncsip módszer
A microarray technológia során elsõ lépésként olyan biológiaicsipeket (2-4 cm2) állítanak elõ (ezek megvásárolhatók, vagy megfelelõ laboratóriumi háttérrel a felhasználó maga állítja elõ), amelyekre rendezetten, sorokban és oszlopokban több ezer, esetleg tízezer jól azonosítható ponton nukleinsavdarabokat (oligonukleotidokat) vagy cDNS (mRNS DNS-re visszaírt formáját) láncokat visznek fel. Felvitelükhöz (mások mellett) az úgynevezett fotolitográfia szolgál, amely során nukleotidonként (szintenként) fényérzékeny védõcsoportokkal ellátott, elõre meghatározott nukleotidokból építik fel a 8-20 elembõl álló láncot. Az eljárás során megfelelõ pontokon átlyuggatott maszkok segítségével alakítják ki a kétdimenziós mintázatot. Ha cDNS-ek felvitele történik, mikrocseppeket alkalmaznak, így kerülnek az ismert szerkezetû cDNS-molekulák a helyükre. A felvitelre használandó felületek továbbfejlesztése jelenleg és a közeljövõben minden bizonnyal tovább emeli az így kialakított elrendezés (array) sûrûségét, tehát az egyidejûleg analizálható gének számát.
E topográfiai információkat és az egyes pontokra felvitt nukleinsavdarab pontos szekvenciáját számítógépben rögzítik. Az így elõkészített array-t inkubálják (hibridizálják) az ismeretlen minta valamilyen színes (például zöld vagy piros) festékkel jelzett nukleinsavdarabjaival. Elõfordul, hogy az ismeretlen minta nukleinsavdarabjait a jelzés elõtt polimeráz láncreakcióval bõvíteni kell. Megfelelõ ionerejû és hõmérsékletû oldattal átmosva õket, csak a részlegesen vagy teljesen azonos nukleotid-komplementeritást mutató darabok maradnak a csip felszínén. Általában a rögzített oligonukleotid közepén található bázisok eltéréseinek kimutatása a legegyszerûbb, itt mosható le legérzékenyebben a mismatch, vagyis a nem tökéletesen illeszkedõ nukleinsavminta. DNS-csipeknél, ahol egy-egy pontmutációra kérdez rá a vizsgáló, ennek megfelelõen tervezik az oligonukleotidokat.
A leolvasás a csip felszínének pásztázó értékelését jelenti, az érzékelõ az egyes pontok színét, intenzitását rögzíti. Ezt követi a bioinformatikai elemzés, tehát a ponthalmaz színelemzése, az adatok szétválogatása és az értékelõ csoportosítása.
Mit vizsgálunk géncsippel?
A DNS-csipeken mutációk (például nukleotidcsere) jelenlétét vagy hiányát keressük. Ez a módszer veleszületett génhibák, illetve az úgynevezett single nucleotide polymorphism (SNP) vizsgálatára használható. Utóbbiak pontmutációk, amelyek mint független genetikai allélok kezelhetõk, kombinált vizsgálatuk individuális genetikai jellegzetességek követését teszi lehetõvé. Több száz vagy ezer SNP egyidejû leolvasása kitûnõen alkalmazható betegségek azonosítására, de akár gyógyszermellékhatások kimutatására is (lásd késõbb). Génexpressziós microarray esetén a kérdés az, hogy két összehasonlítandó mintában (például egy sejt vagy szövet két aktivációs állapota, egy biológiai hatóanyag hatása a kontrollhoz képest, egy egészséges és egy betegbõl származó szövetminta összehasonlítása stb.) milyen gének fejezõdnek ki, és ezek mennyiségileg hogyan viszonyulnak egymáshoz. Ezt génexpressziós profilnak nevezik. A profilok összehasonlításával felismerhetõk és azonosíthatók az adott hatásra jellemzõ gének. Technikailag ez úgy történik, hogy a kétféle mintából származó mRNS-mintát, illetve azok cDNS-re átírt formáját kétféle festék (például zöld és piros) egyikével jelölik, majd keverve a két populációt, hibridizálják azt a mikrocsippel (1. ábra). Amennyiben az egyik (például zöld festékkel jelzett) mintában az adott csipponton reprezentált gén erõsebben fejezõdik ki, a leolvasás itt zöldebb pontot fog detektálni, fordított esetben viszont pirosabb lesz. A legtöbb esetben a két szín megfelelõ kombinációja jelzi a két mintában egy adott gén kifejezõdési arányát. Lehetséges az is, hogy a rögzített expressziós mintázatot ezután adatbankokban már megtalálható, korábban ugyanezen microarray-el nyert kétdimenziós mintázatokhoz hasonlítják, és kiemelik a hasonló eloszlású profilokat. Nyilvánvalóan, a különbözõ hatásokhoz különbözõ génkifejezõdési mintázatok tartoznak. Például a sejtek különbözõ receptorain keresztül kiváltott jelátviteli útjainak génexpressziós mintázatát elemezve, azonosítani lehet ismert szignálok molekuláris következményeit mRNS-szinten. Nagyon lényeges az ebben az összeállításban részletesen nem tárgyalt proteomikai megközelítés is. Ekkor nem cDNS, hanem protein/peptid mintázatok összehasonlítására kerül sor, például kétdimenziós elektroforetogrammal. Immunológiai példával élve: a B-limfocitákon elvégezhetõ az immunglobulin-receptorok, citokinreceptorokon ható jelek, vagy például hisztamininhibitorok hatásának molekuláris követése. Kitûnõen azonosíthatóak a genetikai beavatkozások (például antiszensz hatások, domináns negatív mutáció, génkiütés) hatásai is.
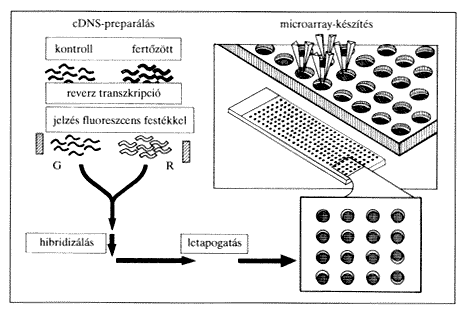
1. ábra. Két különbözõ
mRNS-minta összehasonlítása csippel
Adatbányászás bioinformatika
Az egyes csipekrõl bekerülõ adathalmaz bioinformatikai elemzése nyomán a számítógép clusterekbe rendezi a kapott információkat (2. ábra). Erre egy példa a hierarchiacluster korrelációs analízis. Itt egy olyan mátrix ábrázolás az elemzés eredménye, ahol az egyes oszlopok az egyedi mintákat, az egyes sorok az egyes géneket jelentik. A számítógépes összerendezés lényege az, hogy a hasonló, majd egyre kevésbé hasonló géneket kifejezõ mintákat csoportokba rendezve tüntetik fel. Az egész csip munkalogisztikája tehát úgy fest, hogy az egyes mintákból származó egyedi microarray adatok bioinformatikai összegzése után a számítógépes adatbankok meglévõ mintázatkönyvtárai segítségével következtetéseket vonunk le. A következtetések természete biomedicinális területen legalább kétféle.

2. ábra. A csipanalízis logisztikája
1. Diagnosztika (betegségmegállapítás), prognosztika (betegségkockázat becslése), esetleg megelõzés.
2. Olyan géncsoportok kiemelése, amelyek elõre jeleznek valamilyen például betegségi állapotot, szövõdményt, mellékhatást, tehát van predikciós potenciáljuk.
Ez utóbbi lehetõvé teszi a csip egy második generációjának (diagnózis csip) elõállítását is, amin már csak kizárólag a betegségspecifikus gének szerepelnek. Ez esetenként már csak néhány tucat génnel létrehozandó, nyilvánvalóan olcsóbb makrocsipet igényel.
Ma már lehetséges szövettani blokkokból is rendezett microarray-t készíteni, esetenként akár több száz szövettani blokkból kiemelt hengerek együttes metszése nyomán létrehozott készítményen. Ezen akár nukleinsav-expressziós (például in situ génamplifikáció), akár proteomikai analízis is végezhetõ. Rendelkezésre állnak olyan fixáló reagensek is, amelyek speciálisan protektívak a minta RNS-tartalmát illetõen.
Gyakorlati példák az orvosbiológiából és egy egérmodell
A továbbiakban néhány, a biomedicina területrõl vett példán, és egy kísérleti egérmodellen keresztül mutatom be a génexpressziós vizsgálatokat.
A daganatok. A Bethesda, az MD, az Egyesült Államok sejtbankjaiból 60 különféle emberi daganatos sejtvonal génkifejezõdési mintázatát 1161 gén párhuzamos vizsgálata alapján állapították meg. Ennek során olyan bioinformatikai analízist végeztek, amely kimutatta a különféle szövettani eredetû ráksejtvonalak génexpressziós profiljaiban látható hasonlóságokat és eltéréseket. Ez az adatbank ettõl kezdve összehasonlításként szolgálhat egyedi tumorok jellemzésére.
Egy másfajta bioinformatikai megközelítés a rendelkezésre álló génkifejezõdési adatok alapján olyan halmazokat képez, amelyekbe olyan gének kerülnek, amik a legnagyobb eltérést mutatják a betegség kimenetelében. Így sikerült például melanómában szenvedõ (festékes bõrrák) betegek májáttételét jósló vagy azt nem valószínûsítõ géneket kiemelni. Ezek a gének együttes expressziójuk esetén óriási valószínûséggel mutatják a melanómás beteg várható májáttétét. Ennek korai felismerése nyilvánvalóan gyökeresen más terápiás stratégiát sugall a kezelõorvos számára. Ma már olyan csipek is léteznek, amelyek egy-egy sejt expresszálódó génjeit foglalják magukban, ilyen például a limfocsip, amely az emberi limfociták kifejezõdõ génkészletét tartalmazza. Ennek felhasználásával sikerült például az úgynevezett diffúz, nagy sejtes B-sejtlimfoma betegséget alcsoportokra bontani, amit eddig a hagyományosabb szövettani, immunológiai, sõt molekuláris módszerekkel nem értek el. Az expreszsziós microarray eljárással a betegeket két csoportra osztották: a germinális centrum B, illetve aktivált B jellegû betegekre. Az eredmény jelentõsége az, hogy az elõbbiek betegségkilátásai (például a túlélési idõ) sokszorosan jobbak az utóbbiakénál. Ez megint a kezelési stratégiákban jelent segítséget a klinikus orvosnak.
A fertõzések. A terjedõ és súlyos vírusfertõzések (például a hepatitisz, a HIV) az egyre nagyobb és kiterjedtebb rezisztenciával jellemezhetõ bakteriális és gombafertõzések miatt az egészségügyi (gyógyító és megelõzõ) ellátás világszerte növekvõ kihívás elõtt áll. Központi jelentõsége van a vakcinációnak abban, hogy az egyes fertõzések epidemiológiai következményeit megértsük.
A fertõzésigenomika a mikroorganizmusok és a gazdaszervezet kölcsönhatásának egyedi és fajspecifikus elemeit vizsgálja. Sikerült például a Bordatella alcsoportok eltérõ patogenitása és génexpressziós mintázatai között összefüggést találni.
A szerkezeti és funkcionális genomika ígéretei igen jelentõsek a fertõzésbiológiában. A nem túl távoli jövõben lehetségesnek látszik olyan DNS/cDNS mikrocsip elõállítása, amely potenciálisan minden ma ismert vírus, baktérium és mikroszkopikus gomba összes specifikus génjét tartalmazza. Lehetségesnek látszik tehát a mikrobiológiai diagnosztika felgyorsulása, és az is, hogy a megfelelõ rezisztenciagének kimutatásával például az antibiotikum-érzékenység az eddigieknél sokkal gyorsabban derüljön ki. Hatalmas lehetõség a vakcinakészítés genomikai megközelítése, a mikroorganizmusból a megfelelõ immunológiai céltáblák (epitópok) hatékony és gyors kiemelése, sõt a beteg egyéni genetikai jellegzetességeihez igazított egyéni vakcinatervezés is.
Az allergia. Korunk egyik terjedõ népbetegsége az allergia, az azonnali túlérzékenység és az ehhez kapcsolódó krónikus gyulladási betegségek (például asztma) arányának emelkedése. A környezetbiológiai okok feltárása mellett, úgy látszik, a szerkezeti és funkcionális genomika is nagy szerepet játszik a biomedicinális kutatásban és gyakorlatban. A Semmelweis Egyetem Bõrgyógyászati Klinikájával közösen sikerült például új, eddig nem ismert, vagy funkciójukban nem azonosított géneket találni az atopias dermatitis nevû betegségben. Az érintett gének annotációja (a funkció azonosítása) folyamatban van, remélhetõleg akadnak köztük olyanok is, amelyek új gyógyszercélpontokként jelölhetõk ki.
Fontosnak látszik egyes kemokin- (sejtek mozgását szabályozó citokinek) gének genomikai elemzése, hiszen például az eozinofil sejteknek légzõrendszerbe vándorlása az asztmára való hajlam egyik fontos tényezõjének tekinthetõ. A legfontosabb bioaktív mediátorok egyike a hisztamin. Ennek egyik receptorán genomikai módszerekkel sikerült olyan, a jelátviteli funkciót érintõ eltérést találni, amely szerepet játszik mind az allergiában, mind egyéb (tumoros, gasztroenterológiai) folyamatokban.
Sportgenomika. Genomiális szinten sok génrõl derül ki, hogy allélváltozataik nem közömbösek bizonyos fizikai tulajdonságok szempontjából. Ezek genomikai elemzése edzéstervek módosítását teheti lehetõvé. Expressziós csipek felhasználásával az izomszövet öregedésének jellegzetességeit is sikerült jellemezni.
Gyógyszerkutatás. A farmakogenomika a genomikai analíziseknek az egyik sikerágazata. A már említett SNP-csipek révén mellékhatás jóslása is végezhetõ. A vizsgálat úgy történik, hogy egy adott betegségben (például Alzheimer-kór) kiválasztanak egy adott gyógyszerre mellékhatást mutató és nem mutató csoportot, majd az SNP-csipek eredményeinek bioinformatikai elemzésével keresnek olyan SNP-mintázatot, amely az egyik, vagy a másik csoportra jellemzõ. Ezután, a következõ betegen már csak az SNP-profilt kell meghatározni. Ebbõl már nagy eséllyel megjósolható, hogyan hat a betegre az adott gyógyszer. Ez a technika az egyénre szabott gyógyszeradagolás mellett hosszabb távon még a gyógyszergyártás költségeit is csökkenteni fogja, hiszen sok hatóanyagot nem kell kidobni, csak a megfelelõ célcsoportot kell hozzá megkeresni.
A farmakogenomika másik sikeres iránya a gyógyszer-metabolizációt feltáró (például Cytp450) fehérjék vizsgálata.
Az egérmodell. A génmodifikált egerek fenotípusának vizsgálatakor nagy segítséget nyújthat a genomikai elemzés. Mi hisztamintermelésre képtelen egerek (hisztidin-dekarboxiláz knock-out) embrióit vizsgáltuk három különbözõ expressziós csippel. Munkánk során a normális (vad) és az ugyanolyan törzsbõl származó, de hisztaminmentes egerek embrionális szövetbõl nyert mRNS cDNS formáit használtuk. Mintegy 10 000 egérgént vizsgálva, a gének 5-8 százalékában találtunk változást a gének expressziós mintázatában. A hisztaminhiány miatt megváltozott kifejezõdésû gének azonosítása folyamatban van.
Összegzés
A DNS-microarray géncsiptechnika hatalmas teljesítõképessége, miniatürizálhatósága (nanotechnológia), automatizálhatósága kitûnõen kapcsolódik az informatikai technológia robbanásszerû fejlõdéséhez és a nemzetközi számítógépes hálózati rendszerekhez. A szerkezeti és funkcionális genomika egy eddig nem létezõ sajátosságot eredményez, az in silico kutatás és alkalmazás lehetõségét. Ez az eljárás közvetlen laboratóriumi munka nélkül is jelentõsen kiterjeszti a korszerû tudományt végzõ biológus/orvos/bioinformatikusok körét, tehát térben és idõben növeli a kreatív kutatási tevékenységet végzõ kutatók és alkalmazók számát.

3. ábra. Perspektíva a microarray (csip) analízisben
Biomedicinális értelemben ezzel az új eljárással emberi gének nem ismert funkcióinak azonosítása, új biomarkerek (diagnosztika), potenciális targetek (gyógyszerfejlesztés) kijelölése válik lehetõvé (3. ábra). A genomiális medicina differenciál-diagnosztikai lehetõségekkel, a megelõzés új alternatíváival, a kezelések, gyógyszerhatások-mellékhatások feltárásával gazdagítja az kutatói, klinikai tevékenységet.
| Természet Világa, | 133. évfolyam, 2. szám, 2002. február
https://www.chemonet.hu/TermVil/ https://www.kfki.hu/chemonet/TermVil/ |
Vissza a tartalomjegyzékhez