Proteomkutatás egy új szakterület születése
A 2001. év minden bizonnyal legnagyobb tudományos szenzációja az volt, amikor a Humangenom Program (becenevén HUGO) és a Celera vezetõ kutatói a Nature-ben, valamint a Science hasábjain közzétették az ember genetikai kódjának csaknem teljes szekvenciáját. A program vezetõ szakemberei az 19912005 közötti idõszakra tervezték a génállomány minden egyes betûjének leolvasását, azonban a legújabb generációt képviselõ elemzõ automaták bevonásával látványosan felgyorsultak az események, és a génállomány teljes meghatározása várhatóan már 2003-ra befejezõdik. A program résztvevõi nem kisebb feladatra vállalkoztak, mint hogy megfejtik az emberi DNS közel hárommilliárd betûbõl álló kémiai kódját, amely az egész ember tervrajzát tartalmazza. Felderítik az összes humán gént és helyüket a kromoszómákon, valamint meghatározzák a gének funkcióit. Ez a grandiózus vállalkozás méreteit tekintve vetekszik az Apollo-programmal, melynek révén az ember eljutott a Holdra. A távlatok valóban fantasztikusak. A gének ismeretével az öröklött betegségek diagnosztizálhatóvá válhatnak és már magzati korban is esély lehet a gyógyításukra; teljesen új stádiumba léphet a gyógyszertervezés, s végül az ember önmagáról szerzett ismeretei is soha nem látott mértékben növekedhetnek.
Most, hogy az ember összes kromoszómáját magában foglaló genetikai információ (genom) géntérképét közzétették, a kutatók figyelme arra irányul, hogy a gének által hordozott információhoz funkciót és fehérjekifejezõdési utakat rendeljenek. A genomprogram eddigi lenyûgözõ teljesítménye mögött még elhomályosul az a fontos tény, hogy a megismert struktúrgének 40 százalékának nem ismerjük a pontos biológiai feladatát. A kísérletek nyomán az is világossá vált, hogy nem tartható tovább az a korábbi elv, mely szerint egy gén egy fehérje kódolásáért felelõs. Példa erre a fehérjeszintézis átírási (transzkripció) szakaszában az mRNS-ek módosulása, valamint a leolvasási (transzlációs) fázis utáni fehérjemódosítás, mely megsokszorozza a génekbõl eredõ funkcionális sokféleséget. Ennek a szinte felmérhetetlen komplexitásnak a felismerése és elemzése vezetett el napjainkban az emberi genom megismerése utáni ún. posztgenom korszak beköszöntéséhez. A kutatók számára az is világossá vált, hogy a fehérjeszintézis lépései után bekövetkezõ szabályozási és kémiai módosítási folyamatok mennyiségi és minõségi jellemzése egy a molekuláris biológia és a fehérjekémia határterületén megjelenõ új tudományterület feladata lesz. Ez viszont új szemlélet térhódítását, és ezzel összhangban egy új tudományterület születésének kezdetét is jelenti. Az új tudományterület pedig a proteomkutatás (proteomics), mely nevét a genomkutatás (genomics) analógiájára kapta. Elfogadott és jól hangzó magyar elnevezés híján egyelõre nevezzük mi is proteomkutatásnak, mely lényegét tekintve egy a fehérjéket megcélzó speciális fehérjekutatási módszer.
A tudományterület születésének körülményeit jól példázza, hogy a humán genom közzétételének idõpontjára idõzítve a program szakemberei bejelentették a Human Proteomika Szervezet (Human Proteome Organisation, röviden HUPO) megalakítását is. A HUPO-t alapítói a HUGO megfelelõjének tekintik. Küldetése az, hogy felhívja a figyelmet és anyagi támogatást szerezzen politikai és gazdasági körökben a gének által kódolt fehérjék elemzésére és feladataik tisztázására. A jól ismert és nagy hatékonyságú, klasszikus fehérjefunkció-kutatási módszerektõl a proteomkutatás szemléletében és eszköztárában is elkülönül. Lényege, hogy a forradalmian átalakult fehérjeanalitikai technikák alkalmazásával a sejt vagy a biológiai objektum teljes fehérjekészletét analizálja. Ez sok esetben egyszerre több ezer fehérje, illetve peptid minõségi és mennyiségi viszonyainak leírását jelenti, minimális mintamennyiségbõl (esetleg egyetlen, vagy néhány sejtbõl) kiindulva. A kapott eredményeket tekintve pedig teljesen új távlatokat kínál; a genomban, vagy a sejtre ható külsõ vagy belsõ körülményekben létrehozott, célzott változtatások, illetve a megbetegedések közvetlen biológiai hatását nagy pontossággal és hatékonyan elemezik.
A proteomkutatás
Minél többet tudunk a gének szerepérõl, annál világosabbá válik, hogy a DNS-ben kódolt információ mind a transzkripció, mind a transzláció során jelentõsen megváltozhat, így igen nagyszámú végtermék keletkezéséhez vezethet. A fehérjeszintézis minden egyes lépését számos folyamat ellenõrzi, illetve szabályozza annak érdekében, hogy megfelelõ funkciójú végtermék keletkezzék. A szabályzó folyamatok közül számos a transzkripció idõbeli eltolódásához vezet, valamint feladatát tekintve más fehérje megjelenését eredményezi. Ezek a jelenségek még inkább alátámasztják azt, hogy a genom, és az éppen megjelenõ mRNS vizsgálata nem elegendõ a fenotípus pillanatnyi jellemzésére. Ezek az ellenõrzõ mechanizmusok végül is a funkcionálisan aktív fehérje-, illetve peptidvégtermékek sokkal nagyobb változatosságát okozzák, mint amire korábban az egy génegy fehérje elv alapján gondolhattunk. Most pedig tekintsük át röviden ezeket a folyamatokat!
A transzkripció szintjén bekövetkezõ módosulások
1. Az RNS alternatív hasítása, melynek során a DNS-rõl készült elsõdleges RNS-rõl különféle mRNS-ek keletkezhetnek.
2. A magból történõ RNS-export szabályozása, melynek során egyes RNS-ek a magban visszamaradnak.
3. Az mRNS-export után a citoplazmán belüli RNS lokalizációjának szabályozása. Ez történhet az mRNS által kódolt szignálfehérje segítségével. Maga az mRNS is tartalmazhat olyan jelet (fõleg a le nem fordítódó 3 végen), amely az mRNS sejten belüli elhelyezkedését szabályozza.
4. Az RNS-szerkesztés folyamata. Ennek során, különbözõ szabályozási okok miatt, az mRNS nukleotidszekvenciája megváltozik. Legtöbbször egyetlen nukleotid épül be vagy hasad ki, de ez teljesen megváltoztatja a kódolt fehérje aminosavsorrendjét és ezáltal funkcióját.
5. A transzláció megkezdésének irányítása is sok lehetõséget kínál.
Amikor e szabályozási folyamatok variációinak eredményeként végül is az mRNS lefordítódik a riboszómán, a közvetlen termék, egy jól meghatározott aminosavsorrenddel rendelkezõ fehérje, illetve peptidlánc lesz, aminek funkcionális viselkedését nemcsak az aminosavsorrend határozza meg, hanem egy sor fehérjemódosítási mechanizmus is, amit poszttranszlációs szabályozásnak szokás nevezni.
A transzlációt követõ módosító folyamatok
A kész fehérjék egyes szakaszai (szignalizációs szakaszok) felelõsek azért, hogy a peptid, a fehérje a sejtorganellumok közötti vándorlási lehetõség közül melyiken halad tovább. Ez szabja meg végül is, hogy a fehérje betölti-e azt a feladatot, amelynek végrehajtására megszületett. A riboszómán történt szintézis után a fehérjéket egy különleges fehérjecsoport veszi pártfogásába (chaperone) és hajtogatja össze (protein folding) arra a végsõ formára, amely a nagyszámú lehetséges forma közül a funkció ellátására alkalmassá teszi az adott aminosav-összetételû molekulát. Bizonyos szabályozó folyamatok, amilyen az acetilálás, megvédik az egyes fehérjéket a polipeptidlánc N-terminálisán bekövetkezõ lánchasítástól, fõleg azokat, amelyek végén arginin található. Az ilyen acetilált aminovéggel rendelkezõ fehérjék a sejtben hosszú ideig mûködhetnek anélkül, hogy áldozatul esnének a fehérjebontó folyamatoknak.
A már kész, összehajtogatott fehérje sok esetben további, nem fehérjetermészetû résszel egészülhet ki és ezáltal nyeri el végleges formáját. Ezen folyamatok közül a szénhidrátrész szabályozott beépülése (glikoziláció) a legismertebb, mely jelentõs funkcióváltozást és módosulást okozhat. Ugyancsak fontosak a kináz- és a foszfatáz-enzimrendszerek okozta módosulások. Ebben az esetben a fehérje/peptid aktivitását foszfátcsoportok felvétele, illetve lehasítása szabja meg.
Más mechanizmusok a sejt anyagcseréjében fontos fehérjék mennyiségét szabályozzák. A folyamat során a fehérjék szelektíven bomlanak le (proteolízis) az úgynevezett ubiquitinfüggõ proteaszomális bontás során. Ennek lényege, hogy az inaktiválásra ítélt fehérjéhez speciális, az egész élõvilágban, és szinte minden sejtben azonos felépítésû jelzõpeptid, az ubiquitin kötõdik. Az ubiquitinnel jelölt fehérje így bekerül abba a fehérjekomplexbe, amelynek belsõ felülete aktív fehérjebontó tevékenységet folytat (proteoszom). Mindezek alapján világosan érzékelhetjük, hogy a felsorolt sokféle transzkripciót és transzlációt követõ szabályozási lehetõség miatt egy gén igen sok funkcióért lehet felelõs. A funkció hordozója a végtermékként a sejtben bizonyos ideig életben maradó mûködõ fehérje. Bármely genetikai, környezeti vagy a sejtek anyagcseréjét érintõ hatás megváltoztathatja az adott idõpontban a sejtben feladatukat betöltõ fehérjék minõségét és mennyiségét ugyanazzal a genetikai háttérrel (genommal) rendelkezõ sejtben. E komplex kép leírásával és lehetõségeinek kiaknázásával foglalkozik a proteomkutatás, mely tehát a teljes pillanatnyi fehérjekészletelemzését tûzi ki célul.
A proteomkutatás módszerei
A proteomkutatás módszertana egyszerû. Az adott pillanatban mennyiségi és minõségi felmérést kell végezni az adott fiziológiás állapotú sejt teljes fehérjeállományáról (proteomjáról) és azt össze kell vetni a kísérletesen manipulált, vagy beteg sejt proteomjával. A különbséget okozó fehérje (vagy fehérjék) mennyiségi és minõségi analízise választ adhat kérdéseinkre. A valóságban a megoldás azért bonyolult, mert több tízezer fehérje vagy peptid egyszerre történõ analízisérõl van szó (1. ábra). Ma a legelterjedtebb módszer az, hogy bizonyos elõzetes elválasztási technikával a több tízezer fehérje, peptid közül kiválasztják azt a nagyobb csoportot (szubproteomot), amelyben az összehasonlítani kívánt peptidek, fehérjék valószínûleg megtalálhatók. Például a membránhoz kötõdõ fehérjéknél detergensekkel választják el a hidrofób fehérjéket.

1. ábra. A fehérjeexpressziótól az azonosításig
A következõ lépésként egy újonnan kifejlesztett, úgynevezett immobilis pH-gradiens elektroforézissel (elektromos térben történõ vándoroltatással) a néhány ezer peptidet tartalmazó kivonat molekuláit izoelektromos pontjukban gyûjtik össze (fókuszálják). Ezt az a korszerû eljárás teszi lehetõvé, hogy az egyes gélszakaszok pH-értékét a gél anyagához kovalensen kötött puffermolekulák alakítják ki. Az izoelektromos pontjukban összegyûlõ peptidek ezért a gél adott szakaszán rögzülnek. Az így elválasztott és rögzített peptidcsoportokat egy, az eredeti futásirányra merõleges újabb elektroforézissel molekulatömegük szerint szétválasztják. Érzékeny festési eljárással ezen a kétdimenziós rendszeren 3000-5000 különálló kis peptid-, illetve fehérjefolt jelenik meg. Megfelelõ körülmények kialakításával és az eljárás standardizálásával a kapott foltok összessége adja a vizsgált minta peptidmintázatát vagy fehérje-ujjlenyomatát. A peptidmintázatot a géntechnológiában elfogadott genom szó analógiájára proteom elnevezéssel illetjük. Az egészséges és beteg sejt proteomjának összehasonlításakor megtörténhet, hogy újabb peptidfoltok jelennek meg, de az is elõfordulhat, hogy egyes foltok eltûnnek. Sõt egyes foltok intenzitása erõsödhet, másoké csökkenhet. Mivel ezek szabad szemmel alig, vagy egyáltalán nem érzékelhetõek, ezért speciális pásztázó berendezéseket fejlesztettek ki a géllapok új és/vagy hiányzó foltjainak pontos megjelölésére. Az eredmények számítógépes elemzésével az egészséges és a beteg sejt peptid/fehérjemintázatában bekövetkezõ legcsekélyebb eltérés is kimutatható. Az elemzés utolsó lépése az újonnan megjelenõ vagy az eredeti sejtben meglevõ, de a beteg sejtmintából hiányzó folt mikromódszerekkel történõ kivágása és a piko- vagy nanogramm mennyiségben jelenlévõ peptid- vagy fehérjemolekula vizsgálata, mely ma a fehérjekémia leghatékonyabb csúcstechnikáival, speciális tömegspektrometriával történik (2. ábra).
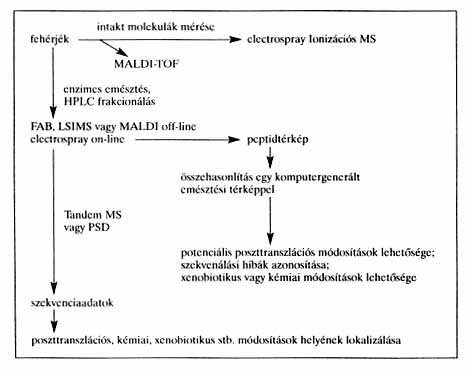
2. ábra. A fehérjeszerkezet analízisének
menete tömegspektrometriával
A tömegspektrometria gyors és rendkívül pontos tömegmérést jelent. A klasszikus tömegspektrometriai mérés során a vizsgálandó anyagot elõször nagy vákuumban elpárologtatják, majd elektronsugárral ionizálják, és az így képzõdött ionokat választják szét elõbb energiájuk szerint (elektrosztatikus analizátor), majd az egységnyi tömegre jutó tömegnek megfelelõen (mágneses analizátor) és az így kétszeresen fókuszált ionokat detektálják. Ez az eljárás még nem tette lehetõvé biológiai minták vizsgálatát, mivel a peptidek, fehérjék túlságosan nagy méretûek és polárisak. Már a minta gõztérbe juttatása is sokáig megoldhatatlan feladatot jelentett. Az utóbbi évtizedben azonban az ugrásszerû fejlõdés eredményeként megjelentek a kíméletes, lágy ionizációs technikák, amelyek lehetõvé tették a hõre érzékeny vagy poláros vegyületek elemzését. Ezeknél az eljárásoknál a vizsgálandó vegyületeket folyadékmátrixban oldják fel és az oldatot vékony rétegben a céltárgy felületére viszik, ahol fókuszált nagy energiájú atom- (xenon) vagy ionsugárral (céziumion) bombázzák. A legújabb fejlesztések eredményeként megjelentek az új típusú ionizációs módszert alkalmazó biológiabarát technikák a tömegspektrometriában (ESI-MS, MALDI-MS), mely utóbbi ionizációs módszert többnyire repülési idõ analizátorral kombinálják. Az egyik ilyen módszernél a fehérjeoldatot, melynek koncentrációja pmol/mikroliter, egy kapillárison keresztül injektálják a tömegspektrométerbe, ahol a belépõ oldatot nagyfeszültség (kb. 4-6 kV) és szárítógáz (nitrogén) fogadja. Töltött cseppeket képeznek, melyekbõl az oldószer elpárolog és visszamarad az analizálandó peptid, vagy fehérje, többszörösen protonált állapotban. Így a fehérje- és peptidmolekulák jellegzetes ionklaszterként jelennek meg a tömegspektrumban, az ionok tömeg/töltés aránya (m/z) függetlenül mérhetõ. Az ionok töltésszáma egyszerû algoritmussal meghatározható és a molekulatömeg többnyire 0,01 százalék pontossággal kiszámítható, azaz egy 100 kDa molekulatömegû fehérje esetében egy aminosav jelenléte vagy hiánya már detektálható. A módszer nagy elõnye, hogy a jó felbontás miatt már viszonylag kis tömegkülönbségek is meghatározhatók.
A másik biológiabarát módszernél (MALDI-TOF) a mátrix olyan vegyület, mely elnyeli a lézer által kibocsátott UV- vagy infrasugárzást és ezzel elõsegíti a benne feloldott fehérjemolekulák ionizálódását. A módszer hihetetlenül érzékeny és akár 1 pikomol minta is elegendõ az analízishez. S végül meg kell említenünk az ismételt, vagy tandem tömegspektrometriát (ahol két MS-készülék van összekapcsolva), mely lehetõvé tette, hogy bõséges szerkezeti információt nyerjünk az elõbb említett lágy ionizációs módszerekkel (FAB, LSIMS, ESIMS) gerjesztett ionokból. Ezek az ionok az elsõ tömegspektrométer után beiktatott ütközési cellában inert gázzal (többnyire hélium vagy argon) ütközve elegendõ energiára tesznek szert, hogy darabokra hulljanak, mely fragmentációs folyamat jellemzõ a molekula szerkezetére. A fragmens ionok a második tömegspektrométer segítségével analizálhatók és detektálhatók.
A tömegspektrométer a fehérjeanalitikában felhasználható az aminosavsorrend meghatározására (szekvenálás) is. Ez viszont azért nagyon fontos, mert a fehérjék primer struktúrájának ismerete alapfeltétele a molekulák térszerkezetének, funkciójának és mûködésük megismerése szempontjából. A fehérjék aminosavsorrendjének közvetlen meghatározására az Edman-lebontáson alapuló eljárások használatosak. Ugyanakkor közvetett módszerek is alkalmazhatók, mint például a kódoló DNS-szakasz bázissorrendjének megállapítása és lefordítása. Ez ugrásszerûen növeli az ismert fehérjeszekvenciákat. Ezek felderítéséhez tehát más módszert kell alkalmazni. Márpedig a szekvenciák pontos meghatározása nélkülözhetetlen. Erre ugyancsak jó lehetõséget kínálnak az elõbb említett biológiabarát technikák, megfelelõ minta-elõkészítést és frakcionálást követõen. A legfejlettebb analitikai rendszerek már több ezer folt aminosav-összetételét is képesek meghatározni naponta. A kapott eredményt az internettel on-line módon összekötött készülékek automatikusan összevetik a nagy nemzetközi adatbázisokban található összetétellel. Ezzel lehetõség nyílik arra, hogy kiderítsék, a szóban forgó kis folt milyen már létezõ szekvenciával azonos, illetve milyen nagy valószínûséggel az. Az összehasonlításból vissza lehet következtetni a peptid/fehérje megfelelõ DNS-szakaszának tulajdonságaira.
A következõkben nézzünk néhány példát e fiatal tudományterület kimagasló eredményeire a mikrobiológia, a neurológia és a rákkutatás területérõl. Már csak annak illusztrálására is érdekesek lehetnek a példák, hogy egyáltalán milyen típusú kérdések megválaszolására lehet alkalmas a proteomkutatás.
A proteomkorszak kezdetének az 1995-ös év tekinthetõ, mikor is Fleischman és munkatársai elsõként tették közzé a Haemophilus influenzae mikroorganizmus teljes proteomjának leírását. Ezt az a technikai fejlõdés tette lehetõvé, mely idõközben a fehérje-géltechnikai módszerek és a nagy felbontású, digitális gélpásztázó rendszerek fejlesztése terén történt. Sikerült azt elérni, hogy egy géllapon egyszerre annyi peptid/fehérjemolekula (kb. 4000-6000 peptid/fehérjemolekula, illetve folt) elválasztható legyen nagy biztonsággal, mint amennyi peptid/fehérje molekulája van az egyszerûbb mikroorganizmusoknak. További fontos kérdésként vetõdött fel, hogy a kezelés hatására megjelenõ vagy eltûnõ fehérjefoltok rendszerbe foglalhatók-e valamilyen általános elv segítségével. Van Boegelen és munkatársai (1996) a mikroorganizmusok fehérjéit aszerint csoportosították, hogy azok a környezeti ingerek, stimulusok hordozói (stimulon) vagy a környezeti hatásra adott válaszban szabályozó funkciót (regulon) látnak-e el. Mint ismeretes, a mikroorganizmusok számos rezisztenciafaktort hordoznak, melyek fontos szerepet játszanak túlélési stratégiájukban. A proteomkutatás módszereivel a rezisztenciafaktorok egész sorát sikerült felfedezni. Példaként említhetjük a Candida albicans azolszármazékok elleni (Marichal és mtsai, 1997; White és mtsai 1998), valamint a Streptomyces törzs eritromicin rezisztenciafaktorait (Cash 1999). Szintén a proteomanalízis módszere tette lehetõvé a veszélyes Helicobacter pylori nevû baktérium virulenciájáért felelõs két fehérje (cagA, vacA) felfedezését (Atherton 1998).
Új távlatokat nyithat az a lehetõség, elsõsorban az idegtudományok terén, hogy szövettenyészetbõl vagy biopsziával az élõ páciensbõl származó néhány sejt is elegendõ ahhoz, hogy a sejtek fehérjéinek pillanatnyi minõségi és mennyiségi állapotáról képet kaphassunk. A betegségspecifikus fehérjék proteomkutatási eljárásokkal történõ feltérképezése máris jelentõs eredményeket hozott a központi idegrendszeri betegségek diagnosztikájában. Feltétlenül ki kell emelnünk itt az Alzheimer-kórral kapcsolatos sajátos neurodegeneratív fehérjék megjelenésének kimutatását (Davidson és mtsai 1999). Langen és munkatársai (1999) 180 neuronspecifikus fehérje osztályozását végezték el. Manji és munkatársai (1999) proteomtechnikával tisztázták a mitogén aktivált protein kináz (MAP-kináz) enzim szerepét a neuropeptidek és a neurotranszmitterek hatásmechanizmusának vizsgálatában.
A rák gyógyítása terén is új lehetõségek adódnak a proteomkutatási technikák bevezetésével. Ma már a vezetõ intézetek szinte egyedi diagnózist képesek felállítani a proteomkutatásra alapozott tipizálási módszerek alkalmazásával. Ehhez azonban a betegség osztályozását segítõ, igen nagyszámú jelzõfehérje gyors elemzésére van szükség. Ebbe az irányba tett egyik jelentõs eredménynek tekinthetõ az a munka, melyet Ostergaard és munkatársai (1997) végeztek. A hólyagrákok 150 lehetséges fajtája között állítottak fel rendszert fehérjeujjlenyomat-vizsgálat alapján. Franzén és csapata (1996) a mellrák legkülönfélébb típusait különítették el a proteomelemzés alapján. Hasonló eredmények születtek a tüdõrák, a leukémia, a petefészek- és prosztatarák különbözõ típusait illetõen is. A sor szerencsénkre szépen bõvül a minél gyorsabb és pontosabb diagnózis felállítását segítve.
Általános az a vélemény, hogy a fehérjetermelés összhangban lesz a betegségek kutatásával és új kezelési eljárásokhoz vezet. Amint azt a Jan Humphrey-Smith, a HUPO egyik alapítója állítja: A fehérjéknek központi jelentõsége van a celluláris funkciók és a betegségek folyamatainak megértésében és a proteomika egységes erõfeszítése nélkül a genomika céljai sem valósulnak meg.
Az említett példák alapján bíztató jövõt jósolhatunk a ma még igazán csak szárnyait próbálgató új tudományterületnek. Az ismertetett módszerek és eljárások ugyanis ma még meglehetõsen költségesek, nyugaton sem számítanak rutin eljárásnak. Beletelik egy kis idõbe, míg a proteomanalízis bevonul a korszerû diagnosztika egyre szélesedõ fegyvertárába. A kezdeti lépéseket azonban már megtettük.
A genomika és a proteomika jövõje
Számos kérdés vetõdik fel, melyekre ma még nem tudjuk a választ. Álljon itt néhány kiragadott példa. Hogyan befolyásolják a gének az emlõsök fejlõdését? Melyek az alapvetõ biokémiai folyamatok, amelyek az élethez szükségesek? Rekonstruálható-e az emberi populáció eredete? Létrehozhatók-e szintetikus életformák? Építhetõ-e komputermodell segítségével olyan sejt, melynek minden komponense ismert, minden folyamata követhetõ, és adott stimulus hatására pontosan mérhetõ a rendszerválasz? Megjósolható-e az aminosav-összetétel alapján a fehérje háromdimenziós szerkezete? A genomika és a proteomika együttesen hogyan változtatja meg az orvos betegségmegelõzõ, diagnosztikai és terápiás tevékenységét?
Valószínû, hogy e kérdéseket még számos követi és a még fel nem tett kérdésekre is meglepõ válaszok születnek az elkövetkezõ évtizedekben. Egy azonban biztos: a megszerzett hatalmas ismeret révén egyre közelebb kerülünk önmagunkhoz és ennek nyomán talán jobban el tudjuk fogadni egymást.
Irodalom
1. Norin, M. and Sundström, M.: Structural proteomics: developments
in structure to function predictions. Trends in Biotechnology. 2002, 20:
7984.
2. Rappsilber J. and Mann, M.: What does it mean to identify a protein
in proteomics? Trends in Biochemical Sciences. 2002, 27: 7478.
3. Nakayama, G. R. Proteomics and genomics. Current Opinion in Chemical
Biology. 2002, 6, 916.
4. Aitken, A. and Learmonth, M.: Protein identification by in gel digestion
and mass spectrometric analysis. Mol. Biotechnol. 2002, 20: 9597.