KÉMIA
A kalmodulin és kismolekulás
komplexei
Szerkezet alapú gyógyszertervezés
HARMAT VERONIKA, KARANCSINÉ MENYHÁRD
DÓRA
A gyógyszertervezés a modern biokémia nagy kihívása.
Kémikus szemmel nézve két eltérô megközelítése
létezik ennek a problémának, a ligandum- és
szerkezet alapú gyógyszertervezés. Az elsô a
hatékonynak bizonyult molekulák szerkezetének általánosítását
jelenti, a második a gyógyszermolekulával kölcsönható
fehérje, ezen belül is az aktív hely szerkezetének
ismeretét igényli. Ez utóbbi eljárás
gyakrabban bizonyult sikeresnek, hiszen ha a fehérje- és
a gyógyszermolekula komplexének térszerkezete ismert,
a két molekula kölcsönhatásának részleteit
is lehet tanulmányozni és értelmezni. Ebben nagy segítséget
nyújt az utóbbi évtizedekben egyre inkább rutinszerûvé
váló szerkezetvizsgáló módszerek mellett
a számítógépes modellezés. A fehérjekrisztallográfiába
és a számítógépes modellezésébe
- az e területen leggyakrabban használt két módszerbe
- szeretnénk betekintést nyújtani egy biológiailag
sokszínû, szerkezetében igen dinamikus fehérje,
a kalmodulin példáján.
A kalmodulin (CaM) több mint harmincféle enzim mûködését
befolyásolja, többek között a sejtosztódás,
a -növekedés, a -kiválasztás és az izom-összehúzódás
szabályozásában vesz részt. Ez a kis méretû
(148 aminosavból
álló), kalciumkötô fehérje minden eukarióta
sejtben megtalálható. A CaM különféle enzimekhez
kötôdik, amikor külsô (hormonális) hatásra
Ca2+ áramlik a sejtbe, amit ezáltal aktivál.
A célenzimek szerkezetüket, funkciójukat, a kalmodulinkötô
régióikat is tekintve igen különbözôek.
Röntgendiffrakciós szerkezetvizsgálatok egész
sora derítette fel, hogy van azonban egy általánosítható
tulajdonsága e fehérjék kalmodulinnal képzett
komplexeinek: a komplex a megfelelô enzimek kalmodulinkötô
régióit bázikus amfifil-hélix formájában
tartalmazza. Ez azt jelenti, hogy ezek a fehérje szakaszok olyan
hélixet alkotnak, amelyek egyik oldalán hidrofób (apoláros,
vízzel gyengén kölcsönható) a másik
oldalon pedig pozitív töltésû aminosavak helyezkednek
el. Mivel a CaM gátlásával sokféle folyamat
befolyásolható, gátlószereit a gyógyászat
különbözô területein használják.
A kalmodulin szerkezeti sajátságai
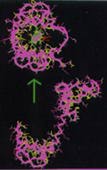 A
CaM - biológiai sokszínûsége mellett - szerkezeti
szempontból is igen érdekes, mert aktiválódása
során meglepôen nagy szerkezeti változáson megy
keresztül. Natív (alap) állapotában rendezetlen
gomolyag szerkezetû, azonban - a már említett Ca2+-beáramlás
hatására súlyzóhoz hasonló formát
vesz fel - ez lesz a fehérje aktív állapota. A CaM
4 Ca2+-iont köt meg. Ezek az ionok a körülöttük
kialakuló oktaéderes koordináció segítségével
kimerevítik a fehérje két terminális régióját:
az eredmény két, egymáshoz egy rendezetlen összekötô
régióval kapcsolt, szinte teljesen azonos egység (domén)
lesz, mely oldószer által könnyen megközelíthetô
felületén hidrofób, körülöttük pedig
savas aminosavak helyezkednek el. A CaM e nagy energiájú
állapotából úgy képes szabadulni, hogy
a közelben tartózkodó enzimek komplementer, tehát
bázikus és hidrofób aminosavakat tartalmazó
szakaszára a két hidrofób doménjével
ráborul, miközben az összekötô régió
meghajlik. A komplexálódás hatása tulajdonképp
ennél határozottabb. A CaM a két hidrofób területtel
mint két hatalmas manccsal megragad, és az enzimbôl
kiemel egy megfelelô - ekkorra már helikális szerkezetû
- szakaszt, amely az enzim belsô inhibítoraként funkcionált:
elfedte az enzim aktív helyét. Ilyen módon aktiválja
a célenzimeit, amelyek aztán különbözô
folyamatok megindulását segítik elô.
A
CaM - biológiai sokszínûsége mellett - szerkezeti
szempontból is igen érdekes, mert aktiválódása
során meglepôen nagy szerkezeti változáson megy
keresztül. Natív (alap) állapotában rendezetlen
gomolyag szerkezetû, azonban - a már említett Ca2+-beáramlás
hatására súlyzóhoz hasonló formát
vesz fel - ez lesz a fehérje aktív állapota. A CaM
4 Ca2+-iont köt meg. Ezek az ionok a körülöttük
kialakuló oktaéderes koordináció segítségével
kimerevítik a fehérje két terminális régióját:
az eredmény két, egymáshoz egy rendezetlen összekötô
régióval kapcsolt, szinte teljesen azonos egység (domén)
lesz, mely oldószer által könnyen megközelíthetô
felületén hidrofób, körülöttük pedig
savas aminosavak helyezkednek el. A CaM e nagy energiájú
állapotából úgy képes szabadulni, hogy
a közelben tartózkodó enzimek komplementer, tehát
bázikus és hidrofób aminosavakat tartalmazó
szakaszára a két hidrofób doménjével
ráborul, miközben az összekötô régió
meghajlik. A komplexálódás hatása tulajdonképp
ennél határozottabb. A CaM a két hidrofób területtel
mint két hatalmas manccsal megragad, és az enzimbôl
kiemel egy megfelelô - ekkorra már helikális szerkezetû
- szakaszt, amely az enzim belsô inhibítoraként funkcionált:
elfedte az enzim aktív helyét. Ilyen módon aktiválja
a célenzimeit, amelyek aztán különbözô
folyamatok megindulását segítik elô.
 |
1. ábra. A Ca2+-kalmodulin
4 TFP (trifluooerazin) molekulával
(szürke) és egy peptiddel (fekete)
alkotott komplexeinek szerkezete.
A fehérjének és a peptidnek
a gerinckonformációja látható,
amit szalagmodellel ábrázoltunk |
A fehérje gátlószerei, a hidrofób és
bázikus részleteket tartalmazó molekulák úgy
hatnak, hogy a Ca2+-aktiválás után kialakult
szerkezet hidrofób zsebeibe kötôdnek a CaM célenzimeivel
versengve (1. ábra).
Módszerek és eredmények
A fehérjék térszerkezetének felderítésére
két kísérleti módszer áll rendelkezésre:
a nukleáris magrezonancia spektroszkópia (NMR) és
a röntgendiffrakció. A CaM mûködése során
bekövetkezô nagymértékû szerkezeti változásokat
mindkét módszerrel behatóan tanulmányozták.
Az NMR spektroszkópia,
amely az atommagok rádiófrekvenciás gerjesztésén
alapuló szerkezetvizsgáló módszer, a molekulák
oldatbeli szerkezetének meghatározására alkalmas.
Hátránya azonban, hogy csak viszonylag kis méretû
fehérjék tanulmányozását teszi lehetôvé.
A röntgendiffrakciós szerkezetvizsgálat alapja, hogy
a röntgensugárzás a vizsgált anyag kristályrácsában,
a rácspontokon elhelyezkedô molekulák elektronfelhôjén
szóródik, és az így nyert diffrakciós
képbôl az elektronsûrûség-függvény
számítható és az atomok helyzete meghatározható.
A molekulaméret növekedése ennél a módszernél
nem okoz gondot, de sok esetben nehézséget jelent a fehérjék
kristályosítása, amely ma is gyakorlatilag próbálgatáson
alapul.
A kristály elektronsûrûség térképe
és a mért diffrakciós kép egymással
bonyolult matematikai viszonyban van, azonban a diffrakciós képbôl
a térszerkezetre jellemzô elektronsûrûség-függvényt
matematikai mûvelettel közvetlenül nem kaphatjuk meg, mert
a mérés során elveszik a szórt sugarak fázisára
vonatkozó információ. Ez a krisztallográfiai
fázisprobléma, melynek megoldására használható
módszerek közül mi a molekuláris helyettesítést
alkalmaztuk, amelynek során egy, az ismeretlen szerkezethez hasonló
ismert szerkezetû modellmolekula (az irodalomban megtalálható
egyik CaM komplex) helyzetét határoztuk meg a kristály
elemi cellájában. Az optimálisan beállított
modellbôl és a mért adatokból számított
függvények átfedése maximális.
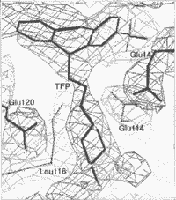
 |
2. ábra. A FTP-molekula
beillesztése a differencia
elektronsûrûségi térképbe.
A differencia elektronsûrûségi
térképet szintfelületes
ábrázolásban szürkével,
a TFP és a fehérje atomjait
feketével ábrázoltuk |
A szerkezetmeghatározás következô lépése
a modellépítés, amelynek során a számítógéppel
grafikusan megjelenített modell konformációját
úgy módosítjuk, hogy illeszkedjen a mért intenzitások,
a fázisok és a modellszerkezet segítségével
számított elektronsûrûségi térképbe.
Ekkor történik a modellben meg nem található
részletek (pl. szubsztrát vagy gyógyszermolekulák)
beillesztése is (2. ábra).
A modellépítés egyes szakaszait szerkezetfinomítási
lépések követik: molekuladinamikai szimuláció
(amelyet a késôbbiekben részletesebben jellemzünk)
segítségével keressük a modellnek azt a konformációját,
amely a mért adatoknak legjobban megfelel, és amellett egyes
atomcsoportokon (aminosavakon) belül az atomok elhelyezkedése
a lehetô legkisebb mértékben tér el a fehérjében
általánosan tapasztalttól.
A röntgendiffrakcióval meghatározott szerkezettel
kapcsolatban gyakran felvetôdik a kérdés, hogy a makromolekulának
a kristályban fellépô többletkölcsönhatások
miatt valamilyen mértékben torzult szerkezete mennyiben tükrözi
a biológiai rendszerekben felvett konformációját,
ez a torzulás azonban legtöbbször elhanyagolható.
A kalmodulint mindkét szerkezetvizsgáló módszerrel
évek óta tanulmányozzák. Meghatározták
a Ca2+-tartalmú fehérje komplexálatlan,
valamint különbözô enzimek kalmodulinkötô
régiójának megfelelô peptidekkel és egy
gyógyszermolekulával, a trifluoperazinnal (TFP, 3.
ábra) alkotott komplexeinek szerkezetét és a Ca2+-mentes
kalmodulin konformációját is. Az NMR-spektroszkópiával
és röntgendiffrakcióval meghatározott szerkezetek
között az egyedüli különbség, hogy az NMR
mérések mutattak rá igazából a Ca2+-CaM
központi összekötô régiójának
nagyfokú oldatfázisbeli rendezetlenségére.
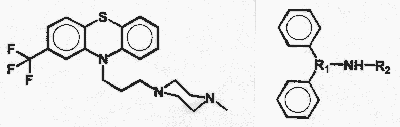
3. ábra. A trifluoperazin (TFP, balra) és difenil-alkil-amin
(DFA) molekulák szerkezeti képlete
A Náray-Szabó Gábor akadémikus vezette csoportunkban
röntgendiffrakciós, valamint számítási
módszerekkel vizsgáljuk a CaM különbözô
típusú gátlószereivel alkotott komplexeit.
(A kristályosítást szegedi kutatókkal együttmûködésben
végezzük.) Célunk a CaM gyógyszermolekulákkal
való kölcsönhatásának jobb megértése,
valamint a ma ismert gyógyszermolekulák hatékonyságát
növelô módosítások javaslata.
A kalmodulin gyógyszermolekulákkal való kölcsönhatását
többféleképpen is el lehet képzelni. Feltételezhetô
egyrészt, hogy a CaM hidrofób felszínén merev
kötôhelyei vannak minden szubsztrát molekulának.
Sikerült CaM-TFP komplexet olyan oldatból kikristályosítanunk,
amely a fehérjét és a gyógyszermolekulát
fiziológiáshoz hasonló arányban tartalmazta.
Az általunk meghatározott szerkezetben a korábban
már felderített 4 TFP-kötôhely közül
csak 2 volt betöltve, de azok a TFP-molekulát az ismerttel
azonos konformációban tartalmazták. Ez az eredmény
rávilágított arra, hogy az eddig feltételezettnél
jóval kevesebb, 4 helyett valószínûleg csak
2 TFP molekula kötôdik a szervezetben a CaM-hoz. Ezen túl
a CaM-2TFP szerkezet bizonyította, hogy a TFP-molekuláknak
igen határozott kötôhelyei vannak a CaM felszínén.
Egy másik gyógyszermolekulával, egy difenil-alkil-amin
származékkal (DFA, 3. ábra) kapcsolatos vizsgálataink
jelentôsen eltérô eredményt szolgáltattak.
A komplex egy kristálymódosulatában az találtuk,
hogy a DFA molekula két különbözô, de egymással
részben átfedô helyzetben kötôdhet a CaM-hoz,
egy másik típusú kristályban (ahol a DFA pontos
elhelyezkedését nem sikerült még meghatásozni)
a fehérje két doménja kicsit más helyzetben
van egymáshoz képest. Tehát míg a TFP-nek jól
meghatározott, különálló kötôhelyei
vannak a CaM-on, a DFA "csúszkáló ligandum",
azaz kötôhelyei, hasonló kötôdési energiával
rendelkeznek (tehát hasonló valószínûséggel
töltôdnek be) és részben átfednek.
Eredményeinket a gyógyszertervezés szemszögébôl
tekintve úgy összegezhetjük, hogy a TFP-molekula hatékonyságának
növelése úgy képzelhetô el, ha kis változtatással
sikerül a protonálható nitrogéneket tartalmazó
részlete és a CaM hidrofób részét határoló
savas aminosavak kölcsönhatását specifikusabbá
tenni. Ezt eredményezhetné a TFP hidrofób részt
és a töltést hordozó gyûrût összekötô
szakaszának meghoszszabbítása. Vizsgálatainkból
az is kitûnik, hogy a gyógyszermolekula hidrofób régióját
is érintô változtatások (lásd DFA) a
kötôhely, a kötési erô és specificitás
megváltozását eredményezhetik, ezért
ilyen típusú módosítások hatása
kísérletek nélkül nehezen felmérhetô.
Számítógépes modellezés
Az eredmények értelmezéséhez és általánosításához
a számítógépes modellezést hívtuk
segítségül.
Egzakt kvantumkémiai számítások a molekulák
mérete miatt kiterjedt biológiai rendszerekre nem alkalmazhatók.
Biomolekulák konformáció analízisére
ma az ún. erôteres módszerek a legelterjedtebbek. E
módszerek a molekulákat merev gömbi atomokból
építik fel, az atomokat rugókkal összekötve.
Így a Hooke-törvény alakjával analóg energiatagok
vezethetôk le.
E = 1/2 k (x-xo)2,
ahol k az erôállandó, x a rugó
nyújtásának mértéke, xo
az egyensúlyi (nyugalmi) rugóhossz. Több ilyen típusú
energiatagot célszerû bevezetni (x: kötéstávolság
-> kötés nyújtási energia, x: kötésszög
-> hajlítási energia, x: torziós szög
-> torziós energia...) amelyek összege a rendszer teljes
energiája. A rugóállandók és az egyensúlyi
geometriához tartozó paraméterek kis molekulákon
elvégzett kvantumkémiai számításokból
vagy kísérleti eredményekként állnak
rendelkezésre. Így a rendszer Hooke-törvény alapján
számított teljes energiája az általunk meghatározott
belsô koordináták (kötéstávolság,
kötésszög, torzió...) sokdimenziós koordinátarendszerében
egy bonyolult potenciálfelszínt alkot, amelynek globális
minimuma a molekula legkedvezoöb és egyúttal legvalószínûbb
konformációját határozza meg. Konformációs
analízisrôl akkor beszélhetünk, ha az általunk
alkalmazott módszer a potenciálfelszín jelentôs
részét "bejárja", azaz képes a felszín
összetettsége miatt létezô nagyszámú
lokális minimum mellett a globális minimum felderítésére
is. Számos ilyen módszer létezik, itt csak az általunk
is használt molekula-dinamika (MD) szimulációk alapelvét
mutatjuk be. A MD-szimulációk során a molekula egyes
atomjaira felírt newtoni mozgásegyenleteket integráljuk
rövid idôközönként. Az egyes atomokra ható
erôbôl számítjuk a megfelelô gyorsulásokat,
amelyeket rövid idôtartamra állandónak tekintve
meghatározzuk az atomok új helyzetét. Ha minden atom
a reá ható erô eredôjének megfelelô
elmozdulását elvégezte, akkor a teljes molekula egy
új konformációja áll elô, a gradiensek
(azaz az erô) újra számíthatók, és
a következô idôlépés alatt bekövetkezô
változás ismét jósolható. A szimulációt
egészen addig folytatjuk, amíg az egy lépés
alatt a rendszerben bekövetkezô változások kicsik
lesznek és elvesztik tendenciózus jellegüket. Ekkor
feltételezhetjük, hogy a globális minimum közvetlen
környezetéhez tartozó konformációk seregét
kapjuk.
A CaM-TFP komplex egy tisztán elméleti úton levezetett
szerkezetét kiindulásul használva MD-szimulációkkal
sikerült reprodukálnunk a TFP legerôsebb kötôhelyére
vonatkozó röntgenkrisztallográfiai eredményeket.
Ennek alapján úgy érezzük, hogy ez a módszer
alkalmas lesz a TFP-molekula esetében javasolt módosítások
hatásának elôzetes tesztelésére, valamint
nehezen vagy egyáltalán nem kristályosítható
komplexek számítógépes tanulmányozására.
| Természet Világa, |
128. évf. 10. sz. 1997. október, 469-471. o. |
|
https://www.kfki.hu/chemonet/TermVil/ |
|
https://www.ch.bme.hu/chemonet/TermVil/ |
Vissza a tartalomjegyzékhez