| KÉMIAI NOBEL-DÍJ, 1998 |
KVANTUMKÉMIA |
FOGARASI GÉZA SZALAY PÉTER
Számítógéppel a molekulák
nyomában
Az 1998. évi Nobel-díjat megosztva John A. Pople és
Walter Kohn kapta a kvantumkémia területén végzett
munkájáért. A kvantumkémia a kémia egyik
ága, mely elméleti alapon, számítógépes
módszerekkel tanulmányozza a molekulákat annak érdekében,
hogy a kémiai tulajdonságokat megértsük, s végsô
célként akár tervezni tudjuk. Ez elvileg mintegy hetven
éve, a kvantummechanika megalkotása óta lehetséges.
Ahhoz azonban, hogy az elvi lehetôség egyre inkább
gyakorlattá válhassék, évtizedek óriási
elméleti-metodikai erôfeszítéseire és
a számítástechnikai forradalomra volt szükség.
A metodikai fejlôdés az elmúlt 3-4 évtizedben
két irányban haladt: a "hagyományos" módszerek
a Y hullámfüggvényre építenek,
míg a sûrûségfunkcionál-elmélet
(Density Functinal Theory, DFT) kísérlet arra, hogy csupán
a jóval egyszerûbb r elektronsûrûséggel
dolgozzunk. E két irány egy-egy kiemelkedô képviselôje
a két kitüntetett, akiknek a következô hivatalos
indoklással adományozták a Nobel-díjat: "Walter
Kohnnak a sûrûségfunkcionál-elmélet kidolgozásáért
és John Pople-nak a kvantumkémiai számítási
módszerek kidolgozásáért."
| John A. Pople 1925-ben született Burnham-on-Sea/Somersetben,
az Egyesült Királyságban. Brit állampolgár.
Matematikából szerzett PhD-fokozatot 1951-ben, Cambridge-ben.
1964-ben a kémiai fizika professzora lett a Carnegie-Mellon Universityn
(Pittsburgh/USA), majd 1986-tól a kémia profeszszora a Northwestern
Universityn (USA), ahol jelenleg is aktív. |
 |
 |
Walter Kohn Bécsben született 1923-ban. 1950 és
1960 között Pittsburghben (USA), a Carnegie Insitute of Technology,
majd 1960-tól 1979-ig San Diegóban, a University of California
professzora. 1979 és 1984 között az Institute of Theoretical
Physics/Santa Barbara igazgatója, ahol ma is dolgozik. |
A kémia alapvetôen kísérleti
tudomány. A vegyész bár számos tapasztalat
alapján nagyon szisztematikusan képes dolgozni s részben
tervezni is a dolog lényegét tekintve mégis szinte
véletlenszerûen, 'trial and error', "próba-szerencse"
elven igyekszik újabb és újabb gyógyszereket,
mûanyagokat, speciális tulajdonságú anyagokat
elôállítani. Jellemzô például,
hogy általában sok száz vagy több ezer vegyületet
kell ahhoz szintetizálni, hogy közülük akár
csak egybôl is gyógyszer lehessen. Felvetôdik a kérdés,
nem lehetne-e ezt a munkát szigorúbb elméleti alapra
helyezni? Az elvi
válasz egyértelmû: a kémiai
tulajdonságokat a molekulákon belüli (intramolekuláris)
és a molekulák közti (intermolekuláris) kölcsönhatások
szabják meg. Ezeknek a természetét pontosan ismerjük,
s igen jó közelitésben egyszerûen a töltött
részecskékre, a pozitív atommagokra, ill. a negatív
elektronokra vonatkozó Coulomb-törvénnyel jellemezhetjük.
A leírás módjára a kvantummechanika kínál
szabatos formalizmust. Ezért írhatta Dirac
már
1929-ben:
"A fizika nagy részének és a kémia
egészének matematikai elméletéhez szükséges
törvények tehát teljes egészében ismertek,
és a nehézség csak abban áll, hogy e törvények
egzakt alkalmazása olyan egyenletekhez vezet, melyek túl
komplikáltak ahhoz, hogy megoldhatók lennének."
A kvantumkémikusok évtizedek óta azon fáradoznak,
hogy sikerüljön az utóbbi problémán túllépni,
s a szóban forgó egyenleteket kellô pontossággal
megoldhatóvá tenni. Ahhoz, hogy e munkák lényegét
megértsük, elôször megpróbáljuk szemléletes,
kvalitatív képben megvilágítani a kémiai
kötés mibenlétét. Ezután vázoljuk
a kvantumkémia fontosabb számítási módszereit,
melyeknek kifejlesztéséhez a most kitüntetett két
tudós jelentôsen hozzájárult. (Érinteni
fogjuk magyar kutatók meghatározó szerepét
is.) Végül néhány példán illusztráljuk,
hol tart ma a kémia ezen ága, melyek a jellemzô alkalmazási
területek.
A kémiai kötés
Közel két évszázada annak, hogy Dalton
igen egyszerû megfigyelések alapján, a kémiai
reakciókban részt vevô anyagok "súlyviszonyainak"
(tömegarányainak) mérésébôl az
anyag atomos szerkezetére következtetett. Az atomok adott rend
szerinti összekapcsolódása a molekula, a kémiai
reakció pedig nem más, mint az atomi kapcsolatok átrendezôdése.
Az a kérdés azonban, hogy milyen erôk tartják
össze az atomokat a molekulákban, rejtély maradt az
egész 19. század során. Nehéz is lett volna
a kémiai kötést megérteni, hiszen az atom közel
egy évszázadig az anyag végsô építôkövét,
megbonthatatlan egységét jelentette a tudomány számára.
A kémiai kötés lényegének megértéséhez
az atom belsô szerkezetének felismerésére volt
szükség. Kerek száz éve, az elektron felfedezése,
majd e század elején az atommag felfedezése szolgáltatta
az atomra vonatkozó mai ismereteink kísérleti alapját:
az atomnak szinte teljes tömege a méretéhez képest
rendkívül tömör atommagban összpontosul, s e
pozitív töltésû mag körül keringenek
a negatív elektronok. Azonban egy ilyen rendszer a klasszikus fizika
szerint képtelenség, nem lehetne stabil, mert a keringô
töltés energiát sugározva be kellene essen a
magba! Az atomi-molekuláris jelenségek leírásához
teljesen új elméletre volt szükség. Az 1920-as
évek közepén megszületett
a kvantummechanika,
a
20. század új világképet jelentô tudományos
eredménye.
A kvantummechanika filozófiai tekintetben is teljesen
új szemléletet vezetett be, melyhez valójában
mindmáig nem könnyû hozzászokni. E szerint a mikrovilágban
a részecskék mozgása elvileg nem követhetô
nyomon olyan részletességgel, ahogyan azt a makrovilágban,
hétköznapi tapasztalataink alapján megszoktuk. Szemben
például a bolygók mozgásával, az atomban
nincs az elektronnak a klasszikus értelemben vett pályája,
mozgásáról csak statisztikus-valószínûségi
kijelentés tehetô. (Ez valóban bizarr szemlélet.
Nem véletlenül berzenkedett Einstein, legalábbis az
értelmezés tekintetében: ,Der liebe Gott würfelt
nicht' "a Jóisten nem szerencsejátékos".)
A kvantummechanika mára teljesen megállapodott,
minden kísérlettel összhangban lévô elmélete
szerint egy rendszer matematikai leírását a Schrödinger-egyenlet
adja:
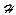 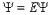 |
(1) |
Ebben a szimbolikusan tömör egyenletben a
,operátor' a Y függvényre
hatva egy bonyolult differenciálegyenletet jelöl ki (E
a rendszer energiája). Az atomot, molekulát a függvény
jellemzi, ennek ismeretében elviekben mindent tudunk a rendszerrôl,
ami a kvantummechanika szerint egyáltalán lehetséges.
Szemléletes tartalma négyzetének van. Ha egyetlen
elektron van az atomban (hidrogénatom), Y2(r)-rel
arányos az elektron megtalálási valószínûsége
a tér egy adott r pontjában. A megtalálási
valószínûséget úgy is értelmezhetjük,
hogy az egyelektronnyi töltés a térben felhôként
oszlik szét és statisztikus átlagban Y2(r)
= r(r)
az elektronsûrûség.
Többelektronos
atomok, molekulák esetében az összes elektronra vonatkozó
információt hordozza, s jelentése bonyolultabb (I.
késôbb). Ilyenkor is jól definiált azonban a
tér egy adott pontjában a teljes, átlagos elektronsûrûség.
 |
1. ábra. A hidrogénmolekula ener-
giája az atomtávolság függvényében |
A kémiai kötés mibenlétét vizsgálhatjuk
energetikai szemlélettel. A kötés ekkor azt jelenti,
hogy a molekulává kapcsolódott
atomok teljes energiája kisebb, mint a szabad atomok energiájának
összege: az atomok molekulává kapcsolódása
energianyereséggel jár. Ez a kérdés a kvantummechanika
segítségével a következõképp ragadható
meg. Tekintsük a legegyszerûbb kétatomos molekulát,
a hidrogént (H2). Ha a magtávolság függvényében
többször megoldjuk az (1) Schrödinger-egyenletet, az energiára
az 1. ábra szerinti minimumgörbét kapjuk. Ezen
"potenciálgödörben" a rendszer stabil. A végtelen
távolságban lévô atomok energiájának
különbsége a gödör mélyének megfelelô
energia, ill. a "disszociációs energia", De. Az
ábrához a számításokat csak demonstrációs
céllal, de valójában igen pontos, "korrelációs"
módszerrel (I. késôbb) végeztük. De
számított értéke 109,1 kcal/mol, a kísérleti
érték 109,4 kcal/mol. (Az egyezés gyakorlatilag tökéletessé
is tehetô.)
Fentieken túl szeretnénk szemléletesen is megérteni,
hogy mi tartja össze a molekulát. Errôl a kvantummechanika
alapján a következô mondható. A molekula ,vázát'
tehát adott (legkedvezôbb) térbeli ekendezôdésben
az atommagok alkotják, s ezen pozitív töltések
terében mozognak az elektronok. Az eredô átlagos
elektronsûrûség túlnyomó része
az atommagokon koncentrálódik, azoknak a töltését
nagyrészt leárnyékolva. A túl egyszerû
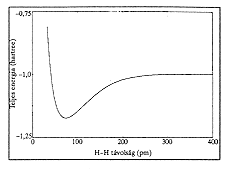
H2-nél kicsit bonyolultabb példát választva,
a 2.a ábra a metil-alkohol
(CH3OH) molekulában
mutatja az elektronsûrûséget olyan léptékben,
hogy csak a legnagyobb értékek észlelhetôek,
a szén- és az oxigénatomon. A H-atomokon a jóval
kisebb maximumok ebben a léptékben nem láthatók,
s még kevésbé látszik az atomok közti
elektronsûrûség. A kémia szempontjából
azonban az a meghatározó, hogy a teljes elektronsûrûségnek
egy kicsiny, de nem elhanyagolható része az atomok közti
térben
koncentrálódik. A 2.b ábra a metil-alkohol
elektronsûrûségének egy másik fajta, szintvonalas
ábrázolása. A függvényértékeket
úgy választottuk, hogy most már jól látható
az elektronsûrûség az atomok közötti térrészben
is: ez a negatív töltésfelhô ,ragasztja' össze
a pozitív atomokat. Ebben a szemléletes megfogalmazásban
tehát az atomok közti térben felhalmozódó
negatív töltés az az "enyv", mely a pozitív töltésû
magokat összetartja, ez jelenti a kémiai kötést.
 |
 |
2.a ábra. Elektronsûrûség-maximumok
a metil-alkohol molekulában |
2.b ábra. A metil-alkohol molekula elektron-
sûrûségének szintvonalas ábrázolása |
A kvantumkémia módszerei
A korábban szimbolikusan felírt (1) Schrödinger-egyenletet
részletezve, ha N elektron mozog térben rögzített
helyzetû atommagok terében, a Hamilton-operátor:
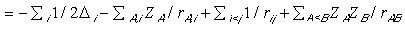 |
(2a) |
és a hullámfüggvény:
| Y = Y(r1,r2,...rN)
= Y(1,2,...N) |
(2b) |
A(2a)-ban Di a második
deriváltakból felépülô Laplace-operátor;
atomi egységeket használtunk, ami azt jelenti, hogy a h
Planck-állandóból
adódó  = h/2p, valamint az elektron tömege
és töltése egység, ezeket nem kellett kiírni;
ZA
az Amag töltése (rendszáma);
rij az
i-edik és j-edik elektron távolsága;
rA,i
az
i-edik elektron távolsága az A magtól,
rAB
az
A és
B mag távolsága. A (2b)-ben ri
az i-edik elektron helykoordinátáit jelöli (az elektronok
spinkoordinátáit az egyszerûség kedvéért
nem jelöltük); a második forma a szokásos tömörebb
jelölés, mely csak indexekkel utal az elektronokra. A szokásos
közelitésben az atommagok rögzítettek, ezért
(2a) utolsó tagja csak egy konstans és a magkoordináták
nem szerepelnek
Y-ben (a magok helyzete csak
egy rögzített paraméter).
= h/2p, valamint az elektron tömege
és töltése egység, ezeket nem kellett kiírni;
ZA
az Amag töltése (rendszáma);
rij az
i-edik és j-edik elektron távolsága;
rA,i
az
i-edik elektron távolsága az A magtól,
rAB
az
A és
B mag távolsága. A (2b)-ben ri
az i-edik elektron helykoordinátáit jelöli (az elektronok
spinkoordinátáit az egyszerûség kedvéért
nem jelöltük); a második forma a szokásos tömörebb
jelölés, mely csak indexekkel utal az elektronokra. A szokásos
közelitésben az atommagok rögzítettek, ezért
(2a) utolsó tagja csak egy konstans és a magkoordináták
nem szerepelnek
Y-ben (a magok helyzete csak
egy rögzített paraméter).
Ebben az általános esetben tehát Y
egy
bonyolult sokváltozós függvény és Y2
jelentése is összetettebb annál, amit az egyelektronos
esetre fentebb elmondtunk. Y2
most
a teljes elektronrendszerre vonatkozóan, az elektronok egy pillanatnyi
elrendezôdését (egy adott elektroneloszlást)
jellemzi. Y2(r1,r2,...)
annak a valószínûségével arányos,
hogy az 1. elektron az r1helyen, ugyanakkor a 2. elektron
az r2 helyen stb. tartózkodik. Ebben a fogalomban
tehát benne van az elektronok mozgásának korrelációja:
az,
hogy milyen valószínûséggel van egy elektron
adott helyen, nem adható meg önmagában, függ attól,
hogy a többi elektron éppen hol van.
A Schrödinger-egyenlet a legegyszerûbb esetektôl
eltekintve nem oldható meg egzakt formában. (Ez a megfogalmazás
sok félreértésre adott okot. Az "egzakt" itt
csak annyit jelent, hogy analitikus függvény formájában
lenne megadható a megoldás. Természetesen ez nem megy,
de a számítógépek korában numerikusan
olyan pontossággal jellemezhetô egy függvény,
hogy az analitikus alak hiánya nem feltétlenül jelent
korlátozást.) Kétségtelen azonban, hogy a kvantumkémia
ma alapvetôen különbözô közelítésekkel
dolgozik.
A hagyományos út: Y. Ahogy
azt a fentiekbôl várjuk, a kvantumkémia szokásos,
"hagyományos" módszerei a Y-függvénnyel
dolgoznak. A legelterjedtebb közelítés, s gyakorlatilag
minden magasabb szintû számításnak is kiindulási
pontja a Hartree- Fock- (HF) módszer. E modell fizikai lényege
abban áll, hogy egy kiszemelt elektron a többi elektron átlagos
terében
mozog. Miként alább megpróbáljuk vázolni,
technikailag ez arra vezet, hogy az általános sokelektronos
Schrödinger-egyenlet helyett csak egy egyelektron egyenletet kell
(igaz sokszor) megoldani. HF-számítások képezik
az eddig végzett sok-sok (bizonyára több százezer)
kvantumkémiai számítás legnagyobb részét
(l. A HarteeFock-módszer).
A HF-modellnek jól megfogható,
szemléletes tartalma van. Az elektronok "pályákon
mozognak", a ji pályán
lévô elektron számára a v potenciál az
a "tér", amelyet a többi elektronnal való átlagos
kölcsönhatásként érez. Fontos azonban
megjegyezni, hogy vHF tartalma elég komplikált,
pontos megértése részletesebb levezetést igényelne.
Hangsúlyozzuk mindenesetre, hogy vHF-ben a "többi
elektronnal való kölcsönhatásban" az ezek által
reprezentált töltéssûrûséggel való
Coulomb-taszítás mellett benne van az ún.
kicserélôdési
effektus is. Ez utóbbi kvalitatíve annyit jelent, hogy
az azonos spinû elektronok (de csak ezek) a determináns
alakú hullámfüggvénybôl kimutatható
módon a Cb-taszításon túlmenôen is
"kerülik" egymást. Ezt hangsúlyozva, az alábbiak
miatt a következô formális szétválasztást
tesszük:
Visszatérve a HF-módszer gyakorlati kérdéseire,
a (4) egyenletet nem differenciálegyenletként
oldják meg, hanem a keresett j pályákat
megfelelôen választott "bázisfüggvényekkel"
(atompályák) sorbafejtve kezelik. A Fock-operátor
szerepét ekkor a "Fock-mátrix" veszi át, s a probléma
a számítógépeknek sokkal jobban megfelelô
"mátrix sajátérték-egyenletre" vezet. Számítástechnikai
szempontból a feladat igen nagyszámú integrál
(a mátrixelemek) kiszámítását és
tárolását, valamint a sajátérték-egyenlet
(többszöri) megoldását jelenti.
Már a fent vázolt legelsô szint, a HF-módszer
sem egyszerû. Nem véletlen, hogy talán két évtizede
sincs, amikor Slater, a kvantumkémia egyik nagy alakja még
elborzadva írt arról, hogy akár több millió
integrál léphet fel a számításokban.
Ezt ô még teljesen járhatatlan útnak tekintette.
Szinte hihetetlen, hogy mára egy HF-számítás
mégis mennyire "rutin" feladat lett. Például a fenti
2.a2.b ábrák a szerzôk által ezen demonstrációs
célra végzett Hartree-Fock számításából
készültek. A metil-alkohol képlete CH3OH,
vagyis 6 atomot tartalmaz, s az elektronok száma 18. Az elvégzett
közepes
ab initio szintû számítás
(melyben közel félmillió integrál lép
fel) mindössze néhány másodpercet igényelt
egy ma átlagos teljesítményûnek nevezhetô
munkaállomáson. HF-szinten ma néhányszor tíz
atomból álló molekulák könnyen számolhatók,
s akár egy-két száz atomig lehet jelenleg felmenni.
Ekkor azonban a gépidô már órákban, napokban
mérhetô.
A HF-módszeren túllépô eljárásokat
"korrelációs
módszerek"-ként szokás emlegetni. Az a célunk,
hogy az elektronok mozgásának fentebb említett korreláltságát
igyekezzünk minél pontosabban figyelembe venni. Az eljárások
technikája különbözô lehet, de nagyjából
mindegyiknek az a lényege, hogy a (3) egyenletbeli
HF egy-determináns helyett determinánsok lineáris
kombinációja a hullámfüggvény:
A determinánsokat úgy állitjuk elô, hogy
a (4) egyenlet megoldásaiból a betöltött
pályák felett adódó, Do-ban betöltetlen
"virtuális"
pályákat
felhasználva, egyes elektronokat magasabb pályákra
teszünk.
A korrelációs számításoknak igen
nagy a számításigénye, s a molekula méretének
(pontosabban: az elektronok számának) magasabb (akár
68-adik!) hatványával nô. A korrelációs
módszerek is különbözô szinteket jelentenek,
a legmagasabb szintû számítások pontossága
már minden igényt kielégít, de ezek 510 atomos
molekulákra korlátozódnak. (Ilyenkor azonban Gbyte
nagyságú központi memória, több tíz
Gbyte diszk-kapacitás szükséges, s a gépidô
a leggyorsabb gépeken is hetekben, akár hónapokban
mérhetô).
A nehézségek ellenére a kvantumkémia e hagyományos
útján óriási fejlôdés volt az
elmúlt 30 évben. Világszerte számos kutatócsoport
és sok száz kutató vett részt az elmélet
fejlesztésében, s e munkák igazi vezéralakja
volt John A. Pople, a Nobel-díj egyik kitüntetettje.
Pople professzor sokoldalú munkájának szintézisét
talán leginkább a Gaussian programcsomag jelképezi.
E számítógép-program elsô verziója
még 1970-ben készült el irányításával.
Mára ebbôl 3040 kutató ,bedolgozásával'
egy kereskedelmi programcsomag lett, mely gyakorlatilag a világ
minden egyetemén és számos kutatóhelyen megtalálható.
A sûrûség funkcionál-elmélet
(DFT). A fent vázolt hagyományos módszer
rendkívüli bonyolultságát látva, kezdetektôl
fogva foglalkoztatta a kutatókat a kérdés: lehetséges-e
egyszerûbb leírás? Miként említettük,
ennek lényege az lenne, hogy a Y hullámfüggvény
helyett a sokkal egyszerûbb r
elektronsûrûségre építsünk elméletet.
Szemben a sokváltozós Y-vel, r
csak a három térkoordináta függvénye,
r=r(r).
Már a kvantummechanika hajnalán voltak ilyen próbálkozások
(Thomas
és
Fermi,
illetve
Dirac),
az
1950-es évektôl pedig
Slater
vezette be az ún.
Xa-módszert. Ezeknek a kísérleteknek
a célja azonban hangsúlyozottan az volt, hogy tovább
egyszerûsítsék a már önmagában is
közelítô HartreeFock-módszert. Új
filozófiát vezetett be e kérdéskörben
a most kitüntetett Walter
Kohn.
Elméleti eredménye
egy nevezetes tétel
(HohenbergKohn-tétel,
1964),
miszerint egy rendszert (atomot, molekulát) egyértelmûen
meghatároz az alapállapotú elektronsûrûség,
r(r).
Utóbbiból tehát meghatározható az energia,
E[r], s ez legalábbis elvileg
nem kell, hogy közelítés legyen: ha E[r]-t
ismernénk, az eredmény egzakt lenne. Ez a sûrûségfunkcionál-elmélet
(DFT)
alapgondolata (lásd A sûrûségfunkcionál-elmélet
technikája).
A DFT-módszer technikája tulajdonképp
visszatérést jelent a szokásos és fent vázolt
HF-módszerhez. A (6) egyenlet teljesen analóg
a (4) egyenlettel, azzal a különbséggel, hogy a Hartree-Fock-potenciál
(4c)-beli vx tagja helyett most a vxc tag szerepel.
Utóbbi a kicserélôdést és a korrelációt
igyekszik egyetlen potenciállal leírni. vxc-re
különbözô formákat használnak, fizikai
modelleken alapuló megfontolások, részben empirikus
tapasztalatok alapján.
A DFT-módszer több évtizedes "vajúdás"
után azzal vált sikeressé, hogy bizonyos technikai
(intégrálási) problémák mellett a
vxc potenciálra a legutóbbi években sikerült
egyre jobb formulákat kidolgozni. Ezáltal az eredmények
meglepôen jók, kb. az egyszerûbb korrelációs
közelítések pontosságát érik el,
azoknál lényegesen ,olcsóbban' (kisebb számítógép-kapacitást
igényelve). Így a DFT-módszer jelenleg a kvantumkémia
nagy "slágere". ADFT gyakorlati sikeréhez számos kutató
járult hozzá, a kitüntetett Kohn professzor érdeme
a tétel megfogalmazása, mely a késôbbi munkákat
inspirálta.
Folytatás
| Természet Világa, |
130. évf. 3. sz. 1999. március, 113119. o.
https://www.kfki.hu/chemonet/TermVil/
https://www.ch.bme.hu/chemonet/TermVil/ |